

Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids
- PMID: 20097765
- PMCID: PMC2856995
- DOI: 10.1074/jbc.M109.092973
Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids
Abstract
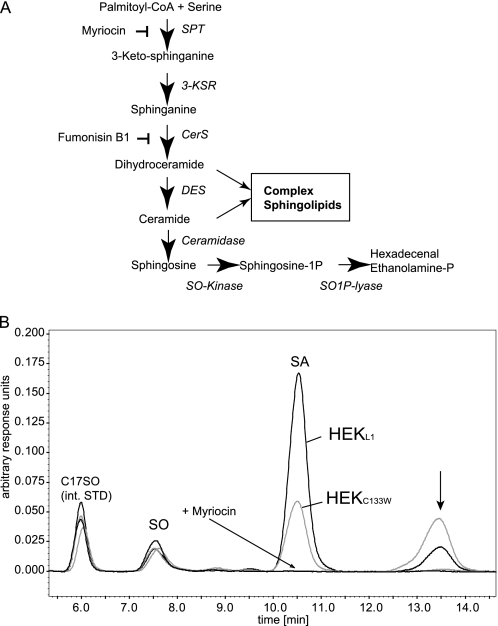
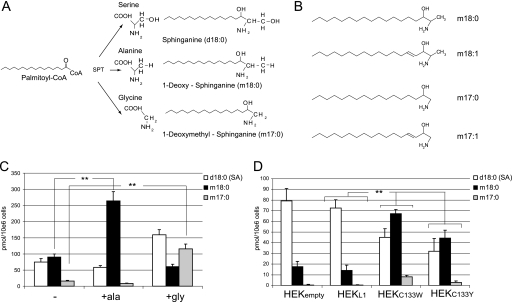
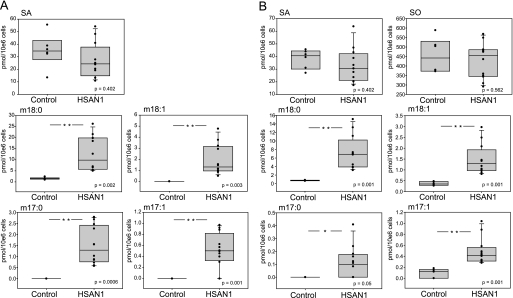
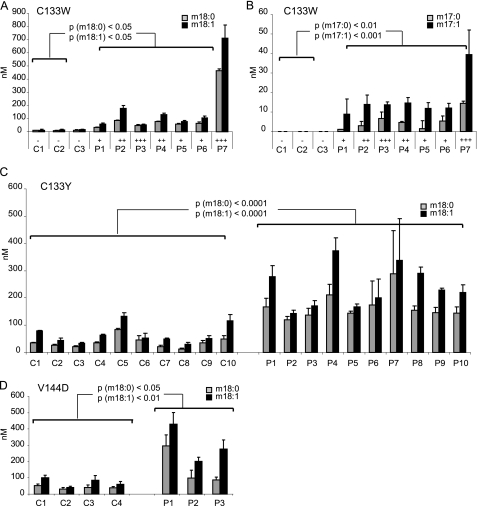
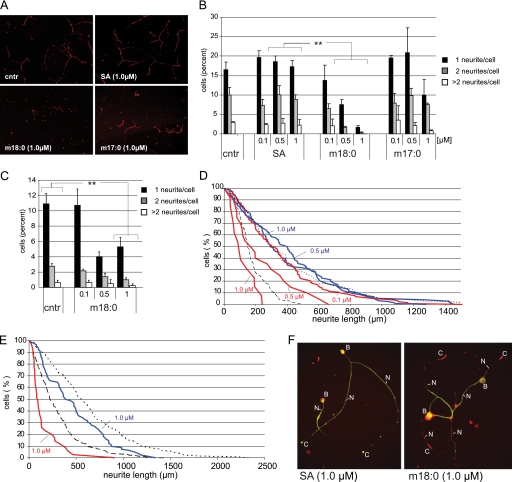
HSAN1 is an inherited neuropathy found to be associated with several missense mutations in the SPTLC1 subunit of serine palmitoyltransferase (SPT). SPT catalyzes the condensation of serine and palmitoyl-CoA, the initial step in the de novo synthesis of sphingolipids. Here we show that the HSAN1 mutations induce a shift in the substrate specificity of SPT, which leads to the formation of the two atypical deoxy-sphingoid bases (DSBs) 1-deoxy-sphinganine and 1-deoxymethyl-sphinganine. Both metabolites lack the C(1) hydroxyl group of sphinganine and can therefore neither be converted to complex sphingolipids nor degraded. Consequently, they accumulate in the cell, as demonstrated in HEK293 cells overexpressing mutant SPTLC1 and lymphoblasts of HSAN1 patients. Elevated DSB levels were also found in the plasma of HSAN1 patients and confirmed in three groups of HSAN1 patients with different SPTLC1 mutations. The DSBs show pronounced neurotoxic effects on neurite formation in cultured sensory neurons. The neurotoxicity co-occurs with a disturbed neurofilament structure in neurites when cultured in the presence of DSBs. Based on these observations, we conclude that HSAN1 is caused by a gain of function mutation, which results in the formation of two atypical and neurotoxic sphingolipid metabolites.
Figures
Similar articles
-
HSAN1 mutations in serine palmitoyltransferase reveal a close structure-function-phenotype relationship.Hum Mol Genet. 2016 Mar 1;25(5):853-65. doi: 10.1093/hmg/ddv611. Epub 2015 Dec 17. Hum Mol Genet. 2016. PMID: 26681808
-
A Novel Variant (Asn177Asp) in SPTLC2 Causing Hereditary Sensory Autonomic Neuropathy Type 1C.Neuromolecular Med. 2019 Jun;21(2):182-191. doi: 10.1007/s12017-019-08534-w. Epub 2019 Apr 6. Neuromolecular Med. 2019. PMID: 30955194
-
Overexpression of the wild-type SPT1 subunit lowers desoxysphingolipid levels and rescues the phenotype of HSAN1.J Neurosci. 2009 Nov 18;29(46):14646-51. doi: 10.1523/JNEUROSCI.2536-09.2009. J Neurosci. 2009. PMID: 19923297 Free PMC article.
-
Serine Palmitoyltransferase (SPT)-related Neurodegenerative and Neurodevelopmental Disorders.J Neuromuscul Dis. 2024;11(4):735-747. doi: 10.3233/JND-240014. J Neuromuscul Dis. 2024. PMID: 38788085 Free PMC article. Review.
-
Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism.Biochim Biophys Acta. 2003 Jun 10;1632(1-3):16-30. doi: 10.1016/s1388-1981(03)00059-3. Biochim Biophys Acta. 2003. PMID: 12782147 Review.
Cited by
-
SPTLC2 variants are associated with early-onset ALS and FTD due to aberrant sphingolipid synthesis.Ann Clin Transl Neurol. 2024 Apr;11(4):946-957. doi: 10.1002/acn3.52013. Epub 2024 Feb 5. Ann Clin Transl Neurol. 2024. PMID: 38316966 Free PMC article.
-
Plasma deoxysphingolipids: a novel class of biomarkers for the metabolic syndrome?Diabetologia. 2012 Feb;55(2):421-31. doi: 10.1007/s00125-011-2384-1. Epub 2011 Nov 29. Diabetologia. 2012. PMID: 22124606
-
Sphingosine-1-phosphate receptor 2.FEBS J. 2013 Dec;280(24):6354-66. doi: 10.1111/febs.12446. Epub 2013 Aug 19. FEBS J. 2013. PMID: 23879641 Free PMC article. Review.
-
Juvenile-onset PSAT1-related neuropathy: A milder phenotype of serine deficiency disorder.Front Genet. 2022 Aug 16;13:949038. doi: 10.3389/fgene.2022.949038. eCollection 2022. Front Genet. 2022. PMID: 36061210 Free PMC article.
-
Chemical biology approaches to membrane homeostasis and function.Chimia (Aarau). 2011;65(11):849-52. doi: 10.2533/chimia.2011.849. Chimia (Aarau). 2011. PMID: 22289370 Free PMC article. Review.
References
-
- Auer-Grumbach M., Mauko B., Auer-Grumbach P., Pieber T. R. (2006) Neuromolecular. Med. 8, 147–158 - PubMed
-
- Dawkins J. L., Hulme D. J., Brahmbhatt S. B., Auer-Grumbach M., Nicholson G. A. (2001) Nat. Genet. 27, 309–312 - PubMed
-
- Bejaoui K., Wu C., Scheffler M. D., Haan G., Ashby P., Wu L., de Jong P., Brown R. H., Jr. (2001) Nat. Genet. 27, 261–262 - PubMed
-
- Hanada K., Hara T., Fukasawa M., Yamaji A., Umeda M., Nishijima M. (1998) J. Biol. Chem. 273, 33787–33794 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
