

Recessive mutations in the putative calcium-activated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies
- PMID: 20096397
- PMCID: PMC2820170
- DOI: 10.1016/j.ajhg.2009.12.013
Recessive mutations in the putative calcium-activated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies
Abstract
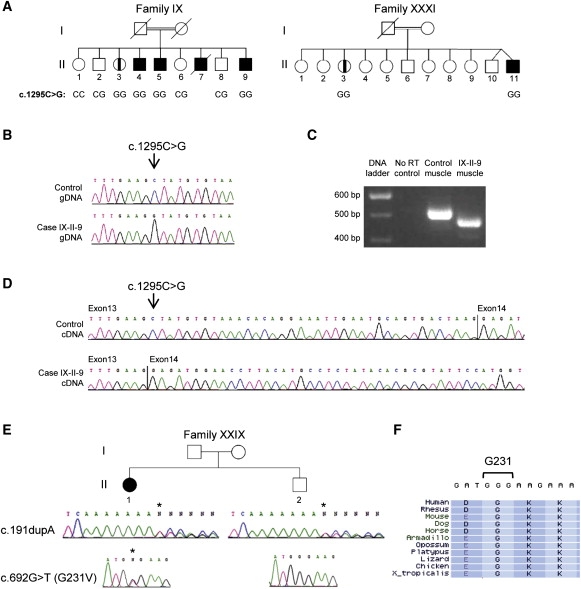
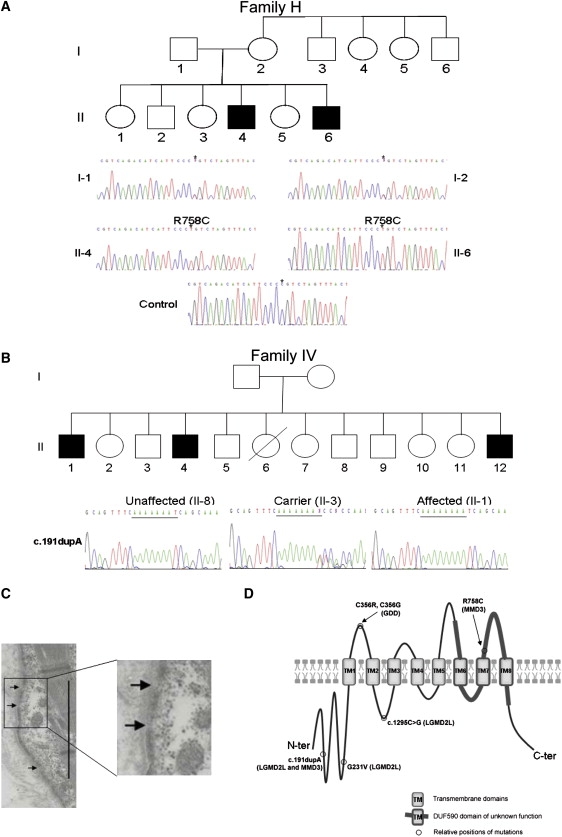
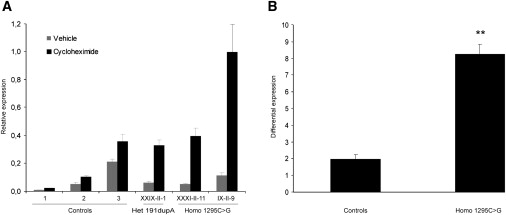
The recently described human anion channel Anoctamin (ANO) protein family comprises at least ten members, many of which have been shown to correspond to calcium-activated chloride channels. To date, the only reported human mutations in this family of genes are dominant mutations in ANO5 (TMEM16E, GDD1) in the rare skeletal disorder gnathodiaphyseal dysplasia. We have identified recessive mutations in ANO5 that result in a proximal limb-girdle muscular dystrophy (LGMD2L) in three French Canadian families and in a distal non-dysferlin Miyoshi myopathy (MMD3) in Dutch and Finnish families. These mutations consist of a splice site, one base pair duplication shared by French Canadian and Dutch cases, and two missense mutations. The splice site and the duplication mutations introduce premature-termination codons and consequently trigger nonsense-mediated mRNA decay, suggesting an underlining loss-of-function mechanism. The LGMD2L phenotype is characterized by proximal weakness, with prominent asymmetrical quadriceps femoris and biceps brachii atrophy. The MMD3 phenotype is associated with distal weakness, of calf muscles in particular. With the use of electron microscopy, multifocal sarcolemmal lesions were observed in both phenotypes. The phenotypic heterogeneity associated with ANO5 mutations is reminiscent of that observed with Dysferlin (DYSF) mutations that can cause both LGMD2B and Miyoshi myopathy (MMD1). In one MMD3-affected individual, defective membrane repair was documented on fibroblasts by membrane-resealing ability assays, as observed in dysferlinopathies. Though the function of the ANO5 protein is still unknown, its putative calcium-activated chloride channel function may lead to important insights into the role of deficient skeletal muscle membrane repair in muscular dystrophies.
Copyright (c) 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Comparing clinical data and muscle imaging of DYSF and ANO5 related muscular dystrophies.Neuromuscul Disord. 2014 Dec;24(12):1097-102. doi: 10.1016/j.nmd.2014.07.004. Epub 2014 Aug 1. Neuromuscul Disord. 2014. PMID: 25176504
-
A founder mutation in Anoctamin 5 is a major cause of limb-girdle muscular dystrophy.Brain. 2011 Jan;134(Pt 1):171-182. doi: 10.1093/brain/awq294. Brain. 2011. PMID: 21186264 Free PMC article.
-
Anoctamin 5 Knockout Mouse Model Recapitulates LGMD2L Muscle Pathology and Offers Insight Into in vivo Functional Deficits.J Neuromuscul Dis. 2021;8(s2):S243-S255. doi: 10.3233/JND-210720. J Neuromuscul Dis. 2021. PMID: 34633328 Free PMC article.
-
Anoctamin 5 (ANO5) Muscle Disorders: A Narrative Review.Genes (Basel). 2022 Sep 27;13(10):1736. doi: 10.3390/genes13101736. Genes (Basel). 2022. PMID: 36292621 Free PMC article. Review.
-
Dysferlinopathies: Clinical and genetic variability.Clin Genet. 2022 Dec;102(6):465-473. doi: 10.1111/cge.14216. Epub 2022 Sep 6. Clin Genet. 2022. PMID: 36029111 Review.
Cited by
-
The ties that bind: functional clusters in limb-girdle muscular dystrophy.Skelet Muscle. 2020 Jul 29;10(1):22. doi: 10.1186/s13395-020-00240-7. Skelet Muscle. 2020. PMID: 32727611 Free PMC article. Review.
-
Muscle Progenitor Cell Fusion in the Maintenance of Skeletal Muscle.Results Probl Cell Differ. 2024;71:257-279. doi: 10.1007/978-3-031-37936-9_13. Results Probl Cell Differ. 2024. PMID: 37996682
-
Clinical and molecular findings in a cohort of ANO5-related myopathy.Ann Clin Transl Neurol. 2019 Jul;6(7):1225-1238. doi: 10.1002/acn3.50801. Epub 2019 Jun 11. Ann Clin Transl Neurol. 2019. PMID: 31353849 Free PMC article.
-
Efficient identification of novel mutations in patients with limb girdle muscular dystrophy.Neurogenetics. 2010 Oct;11(4):449-55. doi: 10.1007/s10048-010-0250-9. Epub 2010 Jul 13. Neurogenetics. 2010. PMID: 20623375 Free PMC article.
-
Predominant localization of phosphatidylserine at the cytoplasmic leaflet of the ER, and its TMEM16K-dependent redistribution.Proc Natl Acad Sci U S A. 2019 Jul 2;116(27):13368-13373. doi: 10.1073/pnas.1822025116. Epub 2019 Jun 19. Proc Natl Acad Sci U S A. 2019. PMID: 31217287 Free PMC article.
References
-
- Guglieri M., Straub V., Bushby K., Lochmuller H. Limb-girdle muscular dystrophies. Curr. Opin. Neurol. 2008;21:576–584. - PubMed
-
- Norwood F., de Visser M., Eymard B., Lochmuller H., Bushby K. EFNS guideline on diagnosis and management of limb girdle muscular dystrophies. Eur. J. Neurol. 2007;14:1305–1312. - PubMed
-
- Cohn R.D., Campbell K.P. Molecular basis of muscular dystrophies. Muscle Nerve. 2000;23:1456–1471. - PubMed
-
- Allikian M.J., McNally E.M. Processing and assembly of the dystrophin glycoprotein complex. Traffic. 2007;8:177–183. - PubMed
-
- Bansal D., Miyake K., Vogel S.S., Groh S., Chen C.C., Williamson R., McNeil P.L., Campbell K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–172. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
