

Loss-of-function mutations in the PRPS1 gene cause a type of nonsyndromic X-linked sensorineural deafness, DFN2
- PMID: 20021999
- PMCID: PMC2801751
- DOI: 10.1016/j.ajhg.2009.11.015
Loss-of-function mutations in the PRPS1 gene cause a type of nonsyndromic X-linked sensorineural deafness, DFN2
Abstract
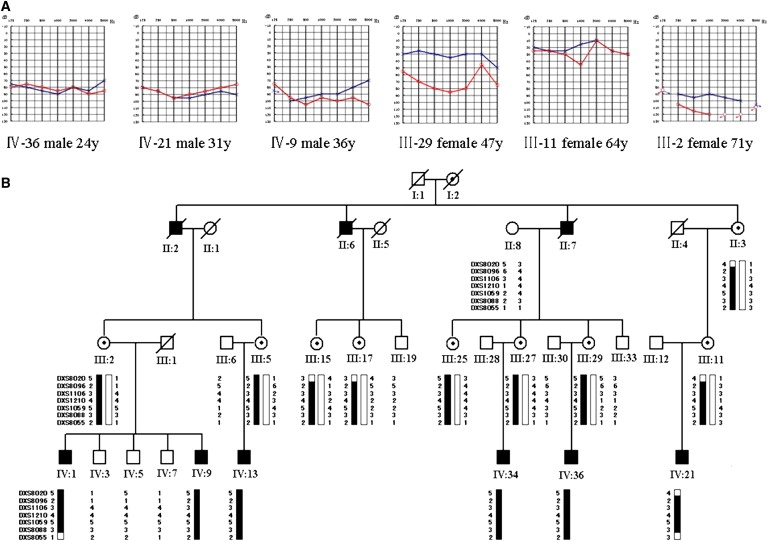
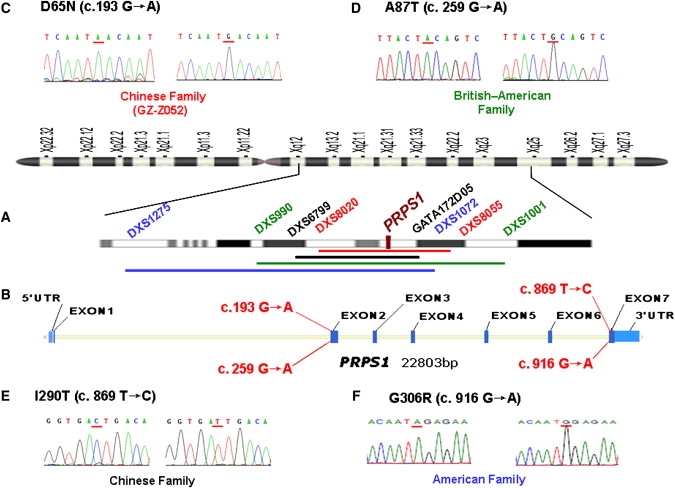
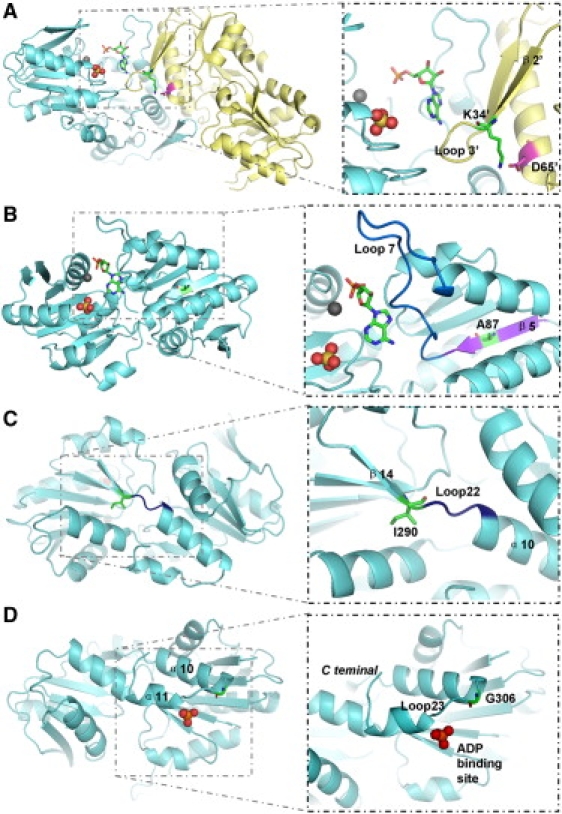
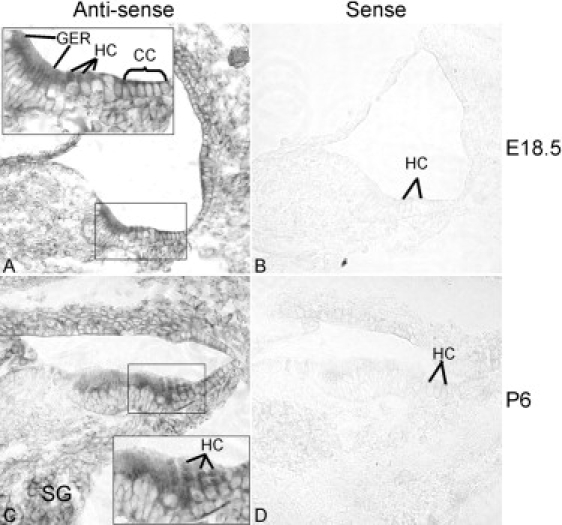
We report a large Chinese family with X-linked postlingual nonsyndromic hearing impairment in which the critical linkage interval spans a genetic distance of 5.41 cM and a physical distance of 15.1 Mb that overlaps the DFN2 locus. Mutation screening of the PRPS1 gene in this family and in the three previously reported DFN2 families identified four different missense mutations in PRPS1. These mutations result in a loss of phosphoribosyl pyrophosphate (PRPP) synthetase 1 activity, as was shown in silico by structural analysis and was shown in vitro by enzymatic activity assays in erythrocytes and fibroblasts from patients. By in situ hybridization, we demonstrate expression of Prps1 in murine vestibular and cochlea hair cells, with continuous expression in hair cells and postnatal expression in the spiral ganglion. Being the second identified gene associated with X-linked nonsyndromic deafness, PRPS1 will be a good candidate gene for genetic testing for X-linked nonsyndromic hearing loss.
2010 The American Society of Human Genetics. Published by Elsevier Inc.
Figures
Similar articles
-
Hearing loss and PRPS1 mutations: Wide spectrum of phenotypes and potential therapy.Int J Audiol. 2013 Jan;52(1):23-8. doi: 10.3109/14992027.2012.736032. Epub 2012 Nov 28. Int J Audiol. 2013. PMID: 23190330 Free PMC article. Review.
-
Functional characterization of a novel loss-of-function mutation of PRPS1 related to early-onset progressive nonsyndromic hearing loss in Koreans (DFNX1): Potential implications on future therapeutic intervention.J Gene Med. 2016 Nov;18(11-12):353-358. doi: 10.1002/jgm.2935. J Gene Med. 2016. PMID: 27886419 Free PMC article.
-
Zebrafish Model for Nonsyndromic X-Linked Sensorineural Deafness, DFNX1.Anat Rec (Hoboken). 2020 Mar;303(3):544-555. doi: 10.1002/ar.24115. Epub 2019 Apr 7. Anat Rec (Hoboken). 2020. PMID: 30874365
-
Mutations in PRPS1 causing syndromic or nonsyndromic hearing impairment: intrafamilial phenotypic variation complicates genetic counseling.Pediatr Res. 2015 Jul;78(1):97-102. doi: 10.1038/pr.2015.56. Epub 2015 Mar 18. Pediatr Res. 2015. PMID: 25785835
-
Association of PRPS1 Mutations with Disease Phenotypes.Dis Markers. 2015;2015:127013. doi: 10.1155/2015/127013. Epub 2015 May 24. Dis Markers. 2015. PMID: 26089585 Free PMC article. Review.
Cited by
-
Novel domain-specific POU3F4 mutations are associated with X-linked deafness: examples from different populations.BMC Med Genet. 2015 Feb 25;16:9. doi: 10.1186/s12881-015-0149-2. BMC Med Genet. 2015. PMID: 25928534 Free PMC article.
-
A novel nonsense CDK5RAP2 mutation in a Somali child with primary microcephaly and sensorineural hearing loss.Am J Med Genet A. 2012 Oct;158A(10):2577-82. doi: 10.1002/ajmg.a.35558. Epub 2012 Aug 10. Am J Med Genet A. 2012. PMID: 22887808 Free PMC article.
-
Genetics of Nonsyndromic Congenital Hearing Loss.Scientifica (Cairo). 2016;2016:7576064. doi: 10.1155/2016/7576064. Epub 2016 Feb 18. Scientifica (Cairo). 2016. PMID: 26989561 Free PMC article. Review.
-
Hearing loss and PRPS1 mutations: Wide spectrum of phenotypes and potential therapy.Int J Audiol. 2013 Jan;52(1):23-8. doi: 10.3109/14992027.2012.736032. Epub 2012 Nov 28. Int J Audiol. 2013. PMID: 23190330 Free PMC article. Review.
-
Contribution of Model Organisms to Investigating the Far-Reaching Consequences of PRPP Metabolism on Human Health and Well-Being.Cells. 2022 Jun 13;11(12):1909. doi: 10.3390/cells11121909. Cells. 2022. PMID: 35741038 Free PMC article. Review.
References
-
- Cohen M.M., Gorlin R.J. Epidemiology, etiology, and genetic patterns. In: Gorlin R.J., Toriello H.V., Cohen M.M., editors. Hereditary hearing loss and its syndromes. Oxford University Press; Oxford: 1995. pp. 9–21.
-
- de Kok Y.J., van der Maarel S.M., Bitner-Glindzicz M., Huber I., Monaco A.P., Malcolm S., Pembrey M.E., Ropers H.H., Cremers F.P. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science. 1995;267:685–688. - PubMed
-
- Tyson J., Bellman S., Newton V., Simpson P., Malcolm S., Pembrey M.E., Bitner-Glindzicz M. Mapping of DFN2 to Xq22. Hum. Mol. Genet. 1996;5:2055–2060. - PubMed
-
- Manolis E.N., Eavey R.D., Sangwatanaroj S., Halpin C., Rosenbaum S., Watkins H., Jarcho J., Seidman C.E., Seidman J.G. Hereditary postlingual sensorineural hearing loss mapping to chromosome Xq21. Am. J. Otol. 1999;20:621–626. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
