

A heterozygous truncating mutation in RRM2B causes autosomal-dominant progressive external ophthalmoplegia with multiple mtDNA deletions
- PMID: 19664747
- PMCID: PMC2725268
- DOI: 10.1016/j.ajhg.2009.07.009
A heterozygous truncating mutation in RRM2B causes autosomal-dominant progressive external ophthalmoplegia with multiple mtDNA deletions
Abstract
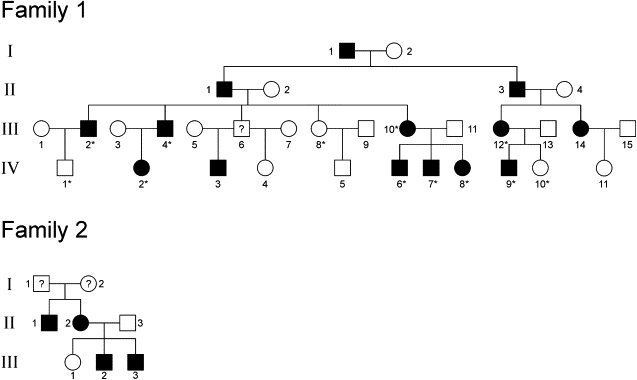
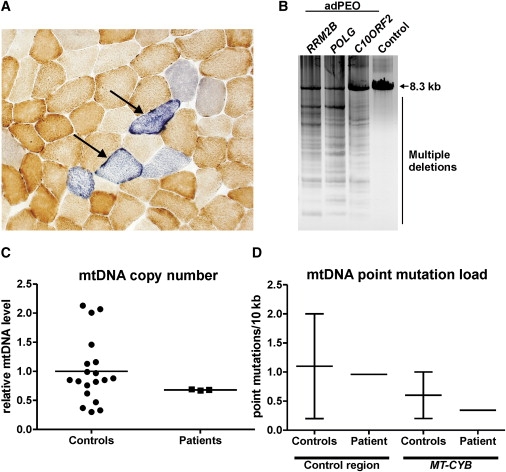
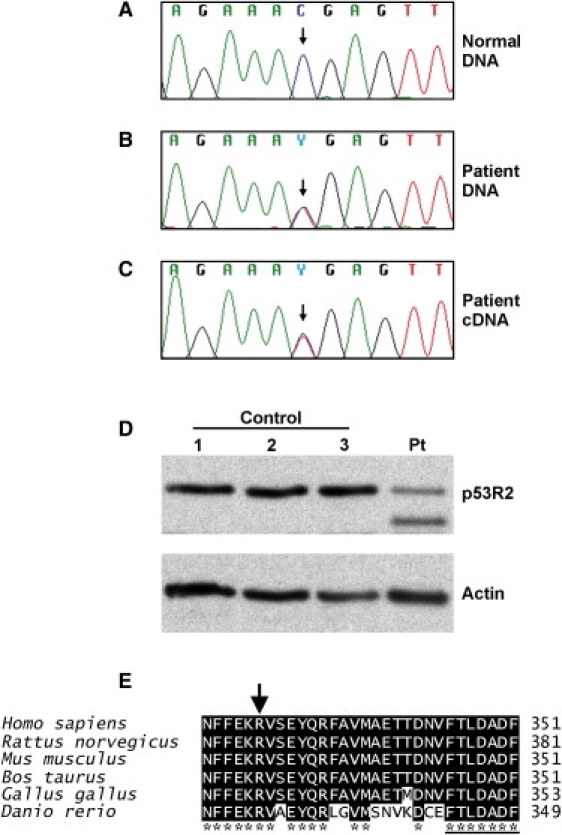
Autosomal-dominant progressive external ophthalmoplegia (adPEO) is a mitochondrial disorder that is characterized by accumulation of multiple mitochondrial DNA (mtDNA) deletions in postmitotic tissues. The disorder is heterogeneous, with five known nuclear disease genes that encode the proteins ANT1, Twinkle, POLG, POLG2, and OPA1. Defects in these proteins affect mtDNA maintenance, probably leading to stalled replication forks, consequent mtDNA deletion formation, and progressive respiratory chain deficiency. Here we present a large adPEO family with multiple mtDNA deletions, whose disease was not explained by mutations in any of the known adPEO loci. We mapped the disease locus in this family to chromosome 8q22.1-q23.3. The critical linkage region contained the RRM2B gene, which encodes the small subunit of the ribonucleotide reductase p53R2, which has previously been shown to be essential for the maintenance of mtDNA copy number. Mutation screening of RRM2B revealed a heterozygous nonsense mutation in exon 9 (c.979C-->T [p.R327X]) in all affected individuals that was absent in 380 control chromosomes. The same mutation was found to segregate in another adPEO family. The mutant mRNA escaped nonsense-mediated decay and resulted in a protein with truncation of 25 highly conserved C-terminal amino acids essential for the interaction with the ribonucleotide reductase subunit R1. We conclude that dominant-negative or gain-of-function mutations in RRM2B are a cause of multiple mtDNA deletions and adPEO.
Figures
Similar articles
-
Progressive external ophthalmoplegia and multiple mitochondrial DNA deletions.Acta Neurol Belg. 2002 Mar;102(1):39-42. Acta Neurol Belg. 2002. PMID: 12094562 Review.
-
Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion.Nat Genet. 2007 Jun;39(6):776-80. doi: 10.1038/ng2040. Epub 2007 May 7. Nat Genet. 2007. PMID: 17486094
-
Exome sequencing identifies a novel missense variant in RRM2B associated with autosomal recessive progressive external ophthalmoplegia.Genome Biol. 2011 Sep 28;12(9):R92. doi: 10.1186/gb-2011-12-9-r92. Genome Biol. 2011. PMID: 21951382 Free PMC article.
-
Kearns-Sayre syndrome caused by defective R1/p53R2 assembly.J Med Genet. 2011 Sep;48(9):610-7. doi: 10.1136/jmg.2010.088328. Epub 2011 Mar 4. J Med Genet. 2011. PMID: 21378381
-
TWINKLE gene mutation: report of a French family with an autosomal dominant progressive external ophthalmoplegia and literature review.Eur J Neurol. 2011 Mar;18(3):436-41. doi: 10.1111/j.1468-1331.2010.03171.x. Epub 2010 Sep 29. Eur J Neurol. 2011. PMID: 20880070 Review.
Cited by
-
Next-generation sequencing reveals DGUOK mutations in adult patients with mitochondrial DNA multiple deletions.Brain. 2012 Nov;135(Pt 11):3404-15. doi: 10.1093/brain/aws258. Epub 2012 Oct 4. Brain. 2012. PMID: 23043144 Free PMC article.
-
The clinical heterogeneity of coenzyme Q10 deficiency results from genotypic differences in the Coq9 gene.EMBO Mol Med. 2015 May;7(5):670-87. doi: 10.15252/emmm.201404632. EMBO Mol Med. 2015. PMID: 25802402 Free PMC article.
-
Identification of a novel heterozygous guanosine monophosphate reductase (GMPR) variant in a patient with a late-onset disorder of mitochondrial DNA maintenance.Clin Genet. 2020 Feb;97(2):276-286. doi: 10.1111/cge.13652. Epub 2019 Nov 14. Clin Genet. 2020. PMID: 31600844 Free PMC article.
-
MPV17 Loss Causes Deoxynucleotide Insufficiency and Slow DNA Replication in Mitochondria.PLoS Genet. 2016 Jan 13;12(1):e1005779. doi: 10.1371/journal.pgen.1005779. eCollection 2016 Jan. PLoS Genet. 2016. PMID: 26760297 Free PMC article.
-
Response to Letter to the Editor: Can MR spectroscopy and muscle biopsy findings be correlated in MELAS and CPEO?CNS Neurosci Ther. 2017 Oct;23(10):848-850. doi: 10.1111/cns.12752. CNS Neurosci Ther. 2017. PMID: 28884978 Free PMC article. No abstract available.
References
-
- Zeviani M., Servidei S., Gellera C., Bertini E., DiMauro S., DiDonato S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature. 1989;339:309–311. - PubMed
-
- Luoma P., Melberg A., Rinne J.O., Kaukonen J.A., Nupponen N.N., Chalmers R.M., Oldfors A., Rautakorpi I., Peltonen L., Majamaa K. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: Clinical and molecular genetic study. Lancet. 2004;364:875–882. - PubMed
-
- Melberg A., Lundberg P.O., Henriksson K.G., Olsson Y., Stalberg E. Muscle-nerve involvement in autosomal dominant progressive external ophthalmoplegia with hypogonadism. Muscle Nerve. 1996;19:751–757. - PubMed
-
- Servidei S., Zeviani M., Manfredi G., Ricci E., Silvestri G., Bertini E., Gellera C., Di Mauro S., Di Donato S., Tonali P. Dominantly inherited mitochondrial myopathy with multiple deletions of mitochondrial DNA: Clinical, morphologic, and biochemical studies. Neurology. 1991;41:1053–1059. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
