

CA8 mutations cause a novel syndrome characterized by ataxia and mild mental retardation with predisposition to quadrupedal gait
- PMID: 19461874
- PMCID: PMC2677160
- DOI: 10.1371/journal.pgen.1000487
CA8 mutations cause a novel syndrome characterized by ataxia and mild mental retardation with predisposition to quadrupedal gait
Abstract
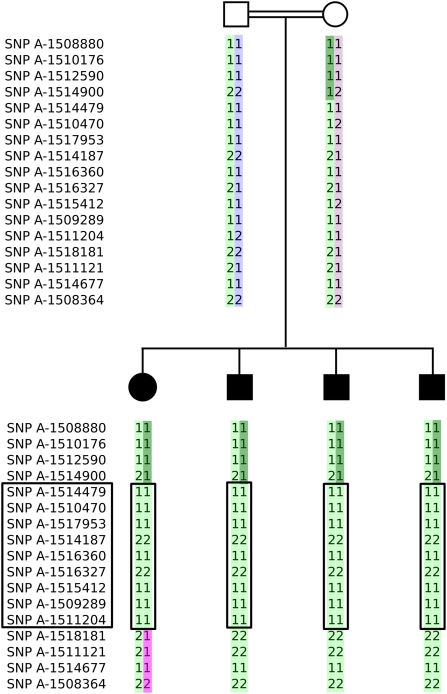
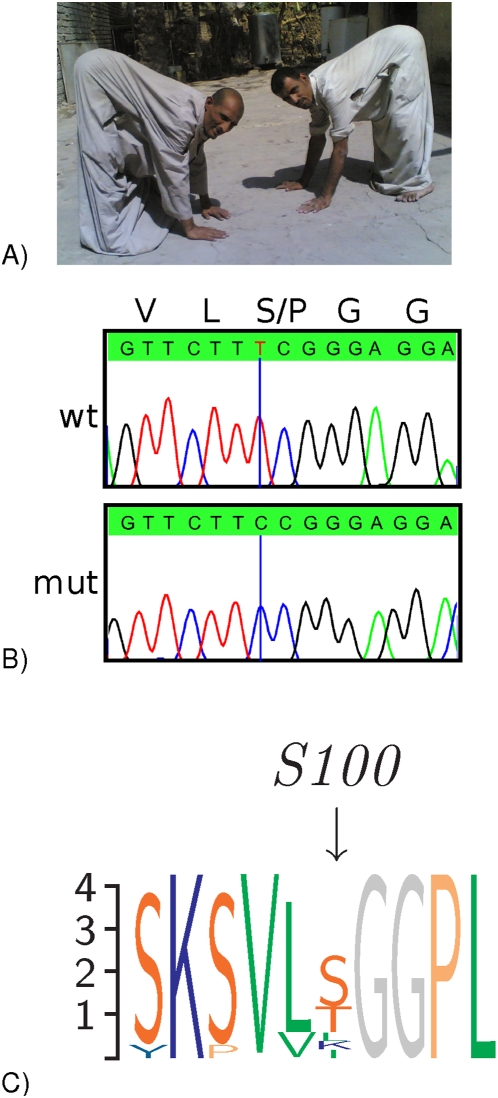
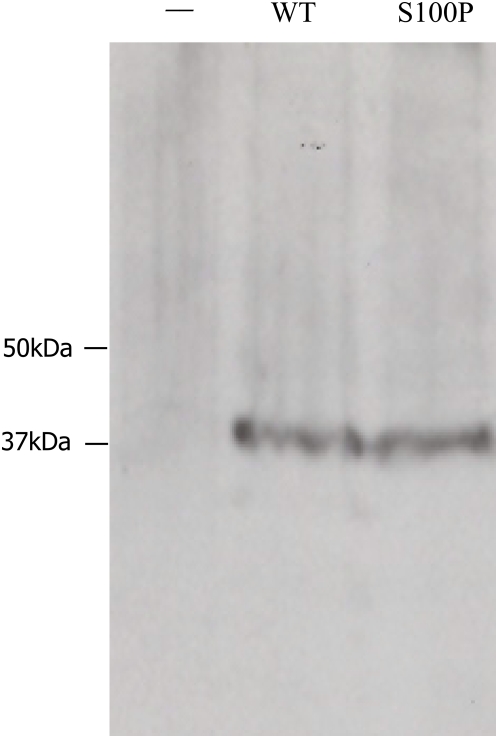
We describe a consanguineous Iraqi family in which affected siblings had mild mental retardation and congenital ataxia characterized by quadrupedal gait. Genome-wide linkage analysis identified a 5.8 Mb interval on chromosome 8q with shared homozygosity among the affected persons. Sequencing of genes contained in the interval revealed a homozygous mutation, S100P, in carbonic anhydrase related protein 8 (CA8), which is highly expressed in cerebellar Purkinje cells and influences inositol triphosphate (ITP) binding to its receptor ITPR1 on the endoplasmatic reticulum and thereby modulates calcium signaling. We demonstrate that the mutation S100P is associated with proteasome-mediated degradation, and thus presumably represents a null mutation comparable to the Ca8 mutation underlying the previously described waddles mouse, which exhibits ataxia and appendicular dystonia. CA8 thus represents the third locus that has been associated with quadrupedal gait in humans, in addition to the VLDLR locus and a locus at chromosome 17p. Our findings underline the importance of ITP-mediated signaling in cerebellar function and provide suggestive evidence that congenital ataxia paired with cerebral dysfunction may, together with unknown contextual factors during development, predispose to quadrupedal gait in humans.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
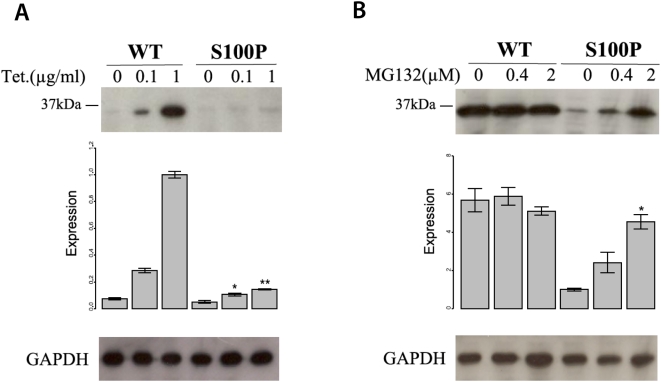
 , **:
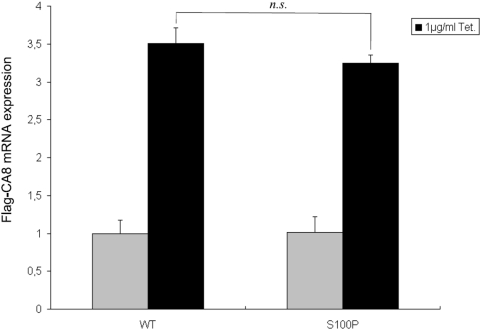
, **:  , comparison between mutant and wildtype at the indicated tetracycline concentration by two-sided t-test. The expression is represented as signal intensity ratio between CA8/GAPDH, which was normalized to the WT level induced by 1 µg/ml tetracycline. Mean values±standard deviation (SD) in triplicate experiments are shown. (B) Rescue of mutant CA8 protein expression by proteasomal inhibition. Addition of the proteasome inhibitor MG132 lead to a dose-dependent rescue of CA8 concentration. *:
, comparison between mutant and wildtype at the indicated tetracycline concentration by two-sided t-test. The expression is represented as signal intensity ratio between CA8/GAPDH, which was normalized to the WT level induced by 1 µg/ml tetracycline. Mean values±standard deviation (SD) in triplicate experiments are shown. (B) Rescue of mutant CA8 protein expression by proteasomal inhibition. Addition of the proteasome inhibitor MG132 lead to a dose-dependent rescue of CA8 concentration. *:  , comparison between 0 µM MG132 and 0.4 µM or 2.0 µM MG132. Expression is shown as ratio relative to that of the mutant CA8 protein (S100P) without MG132. Mean values±SD in triplicate experiments are shown.
, comparison between 0 µM MG132 and 0.4 µM or 2.0 µM MG132. Expression is shown as ratio relative to that of the mutant CA8 protein (S100P) without MG132. Mean values±SD in triplicate experiments are shown.Similar articles
-
Novel homozygous variant of carbonic anhydrase 8 gene expanding the phenotype of cerebellar ataxia, mental retardation, and disequilibrium syndrome subtype 3.Am J Med Genet A. 2020 Nov;182(11):2685-2693. doi: 10.1002/ajmg.a.61805. Epub 2020 Aug 18. Am J Med Genet A. 2020. PMID: 32808436
-
Cerebellar ataxia with normal intellect associated with a homozygous truncating variant in CA8.Clin Genet. 2020 Mar;97(3):516-520. doi: 10.1111/cge.13666. Epub 2019 Nov 14. Clin Genet. 2020. PMID: 31693170
-
A missense founder mutation in VLDLR is associated with Dysequilibrium Syndrome without quadrupedal locomotion.BMC Med Genet. 2012 Sep 14;13:80. doi: 10.1186/1471-2350-13-80. BMC Med Genet. 2012. PMID: 22973972 Free PMC article.
-
Missense mutation in the ITPR1 gene presenting with ataxic cerebral palsy: Description of an affected family and literature review.Neurol Neurochir Pol. 2017 Nov-Dec;51(6):497-500. doi: 10.1016/j.pjnns.2017.06.012. Epub 2017 Jul 8. Neurol Neurochir Pol. 2017. PMID: 28826917 Review.
-
Roles of inositol 1,4,5-trisphosphate receptors in spinocerebellar ataxias.Neurochem Int. 2016 Mar;94:1-8. doi: 10.1016/j.neuint.2016.01.007. Epub 2016 Jan 28. Neurochem Int. 2016. PMID: 26827887 Review.
Cited by
-
Expression and Functional Study of Single Mutations of Carbonic Anhydrase 8 in Neuronal Cells.Cell Mol Neurobiol. 2021 Aug;41(6):1355-1371. doi: 10.1007/s10571-020-00907-1. Epub 2020 Jun 24. Cell Mol Neurobiol. 2021. PMID: 32583043 Free PMC article.
-
Catalytically inactive carbonic anhydrase-related proteins enhance transport of lactate by MCT1.FEBS Open Bio. 2019 Jul;9(7):1204-1211. doi: 10.1002/2211-5463.12647. Epub 2019 Jun 11. FEBS Open Bio. 2019. PMID: 31033227 Free PMC article.
-
Structural Characterization of Carbonic Anhydrase VIII and Effects of Missense Single Nucleotide Variations to Protein Structure and Function.Int J Mol Sci. 2020 Apr 16;21(8):2764. doi: 10.3390/ijms21082764. Int J Mol Sci. 2020. PMID: 32316137 Free PMC article.
-
Current Opinions and Consensus for Studying Tremor in Animal Models.Cerebellum. 2019 Dec;18(6):1036-1063. doi: 10.1007/s12311-019-01037-1. Cerebellum. 2019. PMID: 31124049 Free PMC article. Review.
-
Human carbonic anhydrase-8 AAV8 gene therapy inhibits nerve growth factor signaling producing prolonged analgesia and anti-hyperalgesia in mice.Gene Ther. 2018 Jul;25(4):297-311. doi: 10.1038/s41434-018-0018-7. Epub 2018 Apr 24. Gene Ther. 2018. PMID: 29789638 Free PMC article.
References
-
- Schöls L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3:291–304. - PubMed
-
- Fogel BL, Perlman S. Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet Neurol. 2007;6:245–257. - PubMed
-
- Türkmen S, Hoffmann K, Demirhan O, Aruoba D, Humphrey N, et al. Cerebellar hypoplasia, with quadrupedal locomotion, caused by mutations in the very low-density lipoprotein receptor gene. Eur J Hum Genet. 2008;16:1070–1074. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
