

Osteopetrosis
- PMID: 19232111
- PMCID: PMC2654865
- DOI: 10.1186/1750-1172-4-5
Osteopetrosis
Abstract
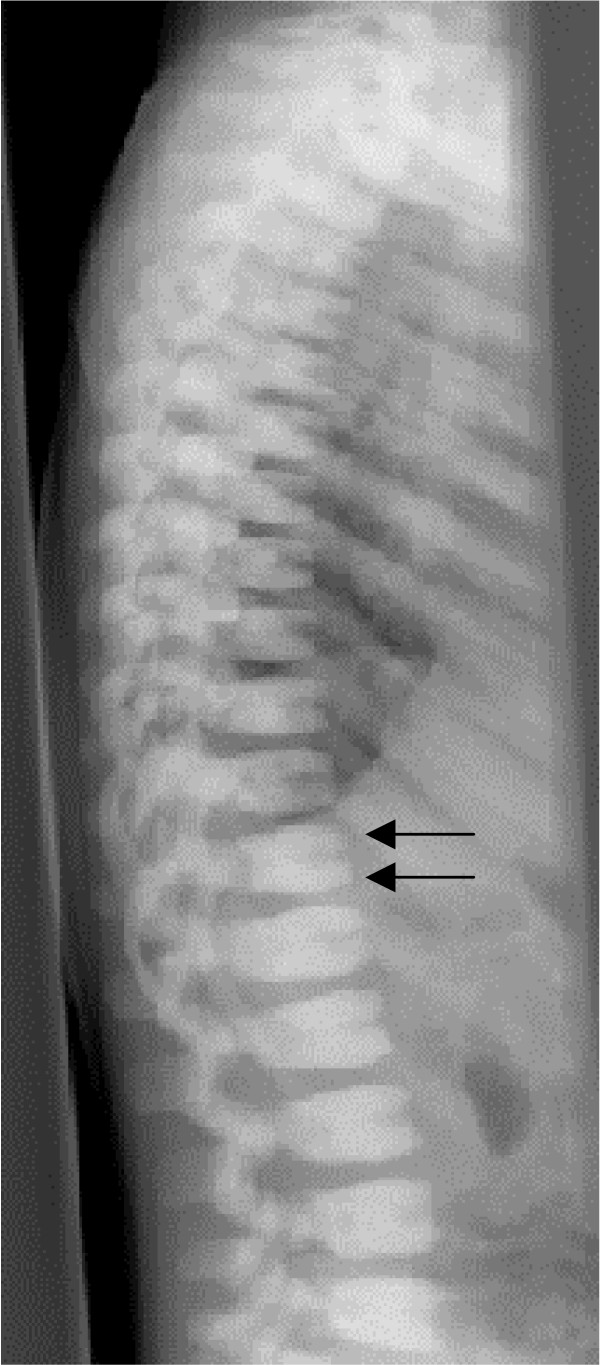
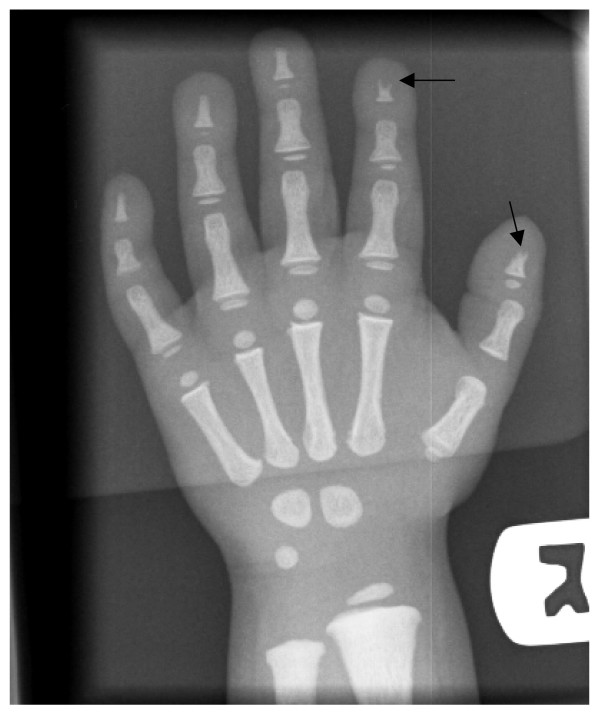
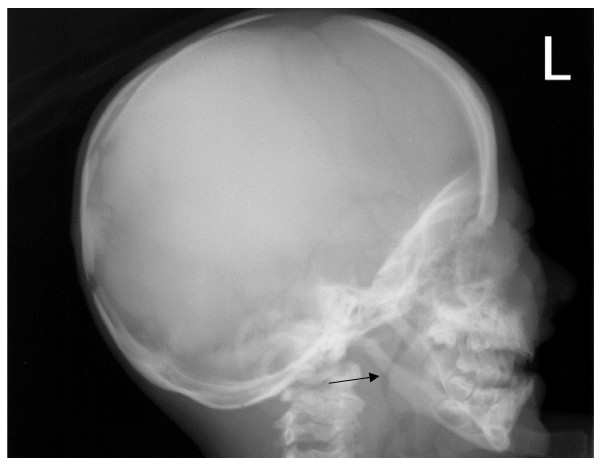
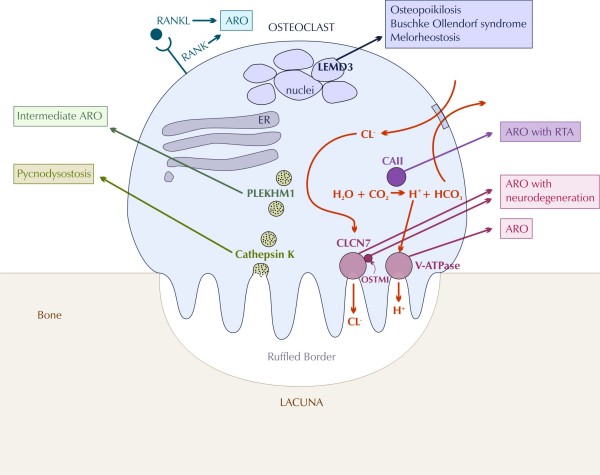
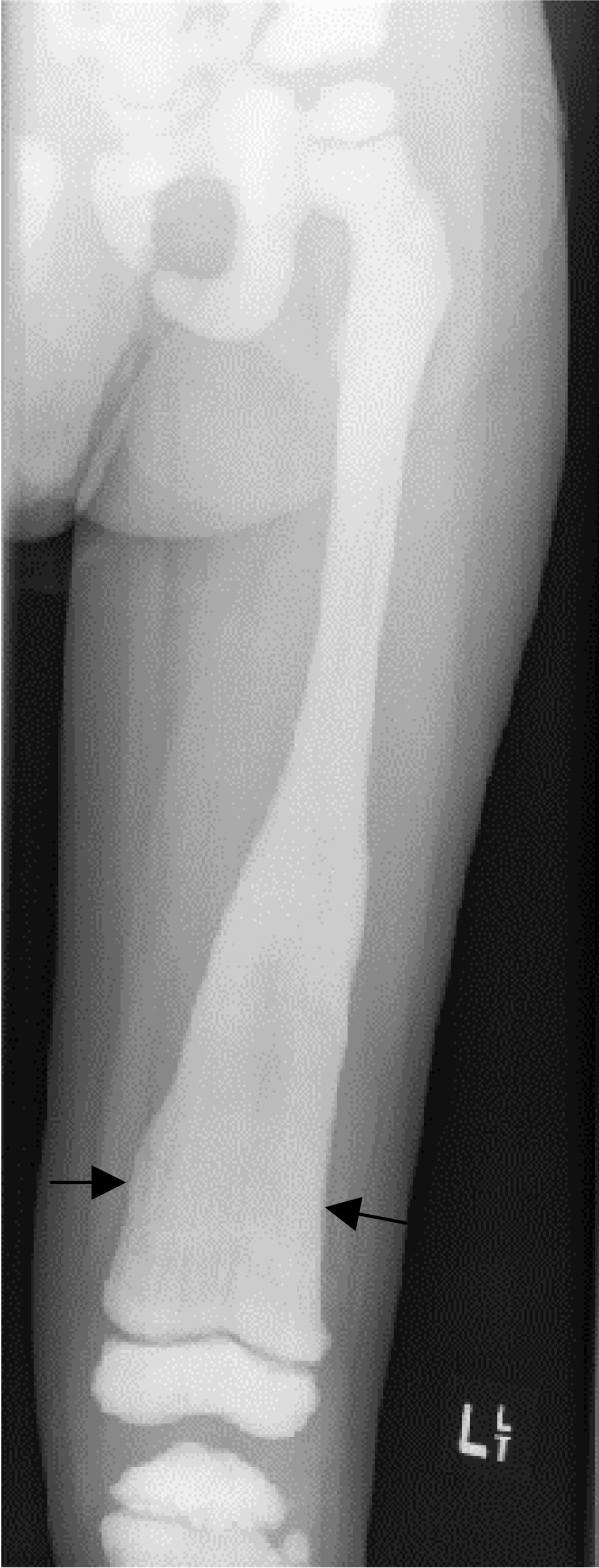
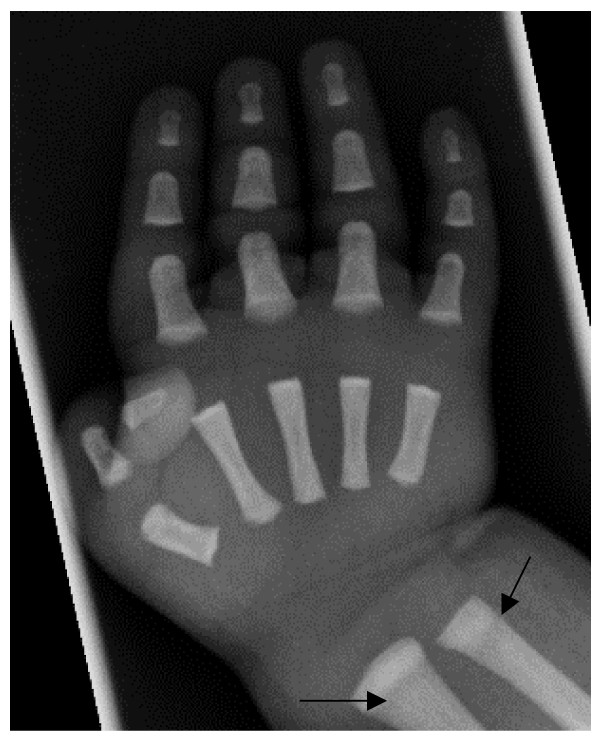
Osteopetrosis ("marble bone disease") is a descriptive term that refers to a group of rare, heritable disorders of the skeleton characterized by increased bone density on radiographs. The overall incidence of these conditions is difficult to estimate but autosomal recessive osteopetrosis (ARO) has an incidence of 1 in 250,000 births, and autosomal dominant osteopetrosis (ADO) has an incidence of 1 in 20,000 births. Osteopetrotic conditions vary greatly in their presentation and severity, ranging from neonatal onset with life-threatening complications such as bone marrow failure (e.g. classic or "malignant" ARO), to the incidental finding of osteopetrosis on radiographs (e.g. osteopoikilosis). Classic ARO is characterised by fractures, short stature, compressive neuropathies, hypocalcaemia with attendant tetanic seizures, and life-threatening pancytopaenia. The presence of primary neurodegeneration, mental retardation, skin and immune system involvement, or renal tubular acidosis may point to rarer osteopetrosis variants, whereas onset of primarily skeletal manifestations such as fractures and osteomyelitis in late childhood or adolescence is typical of ADO. Osteopetrosis is caused by failure of osteoclast development or function and mutations in at least 10 genes have been identified as causative in humans, accounting for 70% of all cases. These conditions can be inherited as autosomal recessive, dominant or X-linked traits with the most severe forms being autosomal recessive. Diagnosis is largely based on clinical and radiographic evaluation, confirmed by gene testing where applicable, and paves the way to understanding natural history, specific treatment where available, counselling regarding recurrence risks, and prenatal diagnosis in severe forms. Treatment of osteopetrotic conditions is largely symptomatic, although haematopoietic stem cell transplantation is employed for the most severe forms associated with bone marrow failure and currently offers the best chance of longer-term survival in this group. The severe infantile forms of osteopetrosis are associated with diminished life expectancy, with most untreated children dying in the first decade as a complication of bone marrow suppression. Life expectancy in the adult onset forms is normal. It is anticipated that further understanding of the molecular pathogenesis of these conditions will reveal new targets for pharmacotherapy.
Figures
Similar articles
-
Autosomal dominant osteopetrosis: clinical severity and natural history of 94 subjects with a chloride channel 7 gene mutation.J Clin Endocrinol Metab. 2007 Mar;92(3):771-8. doi: 10.1210/jc.2006-1986. Epub 2006 Dec 12. J Clin Endocrinol Metab. 2007. PMID: 17164308
-
Autosomal-dominant osteopetrosis: an incidental finding.Indian J Dent Res. 2010 Oct-Dec;21(4):611-4. doi: 10.4103/0970-9290.74234. Indian J Dent Res. 2010. PMID: 21187637 Review.
-
Osteopetrosis and related osteoclast disorders in adults: A review and knowledge gaps On behalf of the European calcified tissue society and ERN BOND.Eur J Med Genet. 2024 Jun;69:104936. doi: 10.1016/j.ejmg.2024.104936. Epub 2024 Apr 7. Eur J Med Genet. 2024. PMID: 38593953 Review.
-
Chloride channel ClCN7 mutations are responsible for severe recessive, dominant, and intermediate osteopetrosis.J Bone Miner Res. 2003 Oct;18(10):1740-7. doi: 10.1359/jbmr.2003.18.10.1740. J Bone Miner Res. 2003. PMID: 14584882
-
Clinical, genetic, and cellular analysis of 49 osteopetrotic patients: implications for diagnosis and treatment.J Med Genet. 2006 Apr;43(4):315-25. doi: 10.1136/jmg.2005.036673. Epub 2005 Aug 23. J Med Genet. 2006. PMID: 16118345 Free PMC article.
Cited by
-
Type 3 renal tubular acidosis.Indian J Nephrol. 2012 Nov;22(6):466-8. doi: 10.4103/0971-4065.106058. Indian J Nephrol. 2012. PMID: 23439805 Free PMC article.
-
Simple reproducible technique in treatment for osteopetrotic fractures.Musculoskelet Surg. 2013 Aug;97(2):117-21. doi: 10.1007/s12306-012-0222-3. Epub 2012 Sep 16. Musculoskelet Surg. 2013. PMID: 22983737
-
A Clinical Perspective on Advanced Developments in Bone Biopsy Assessment in Rare Bone Disorders.Front Endocrinol (Lausanne). 2020 Jun 23;11:399. doi: 10.3389/fendo.2020.00399. eCollection 2020. Front Endocrinol (Lausanne). 2020. PMID: 32714279 Free PMC article.
-
Managing Osteopetrosis in the Complex Polytrauma Orthopedic Patient.Cureus. 2022 Feb 3;14(2):e21886. doi: 10.7759/cureus.21886. eCollection 2022 Feb. Cureus. 2022. PMID: 35265418 Free PMC article.
-
Novel mutation of TCIRG1 and clinical pictures of two infantile malignant osteopetrosis patients.J Bone Miner Metab. 2011 Mar;29(2):251-6. doi: 10.1007/s00774-010-0228-6. Epub 2010 Nov 2. J Bone Miner Metab. 2011. PMID: 21042819
References
-
- Albers-Schonberg Rontgenbilder einer seltenen Knockenerkrankung. Munch Med Wochenschr. 1904;5:365–368.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
