

A critical role for glycine transporters in hyperexcitability disorders
- PMID: 18946534
- PMCID: PMC2526004
- DOI: 10.3389/neuro.02.001.2008
A critical role for glycine transporters in hyperexcitability disorders
Abstract
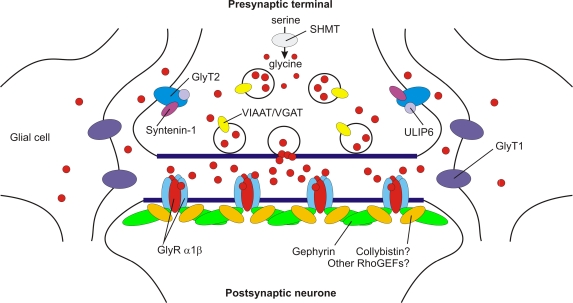
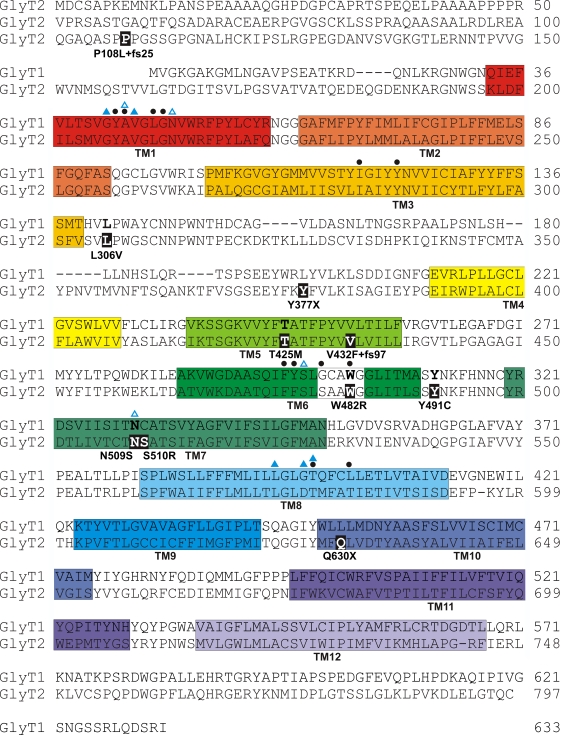
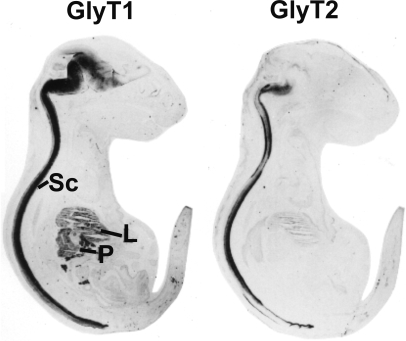
Defects in mammalian glycinergic neurotransmission result in a complex motor disorder characterized by neonatal hypertonia and an exaggerated startle reflex, known as hyperekplexia (OMIM 149400). This affects newborn children and is characterized by noise or touch-induced seizures that result in muscle stiffness and breath-holding episodes. Although rare, this disorder can have serious consequences, including brain damage and/or sudden infant death. The primary cause of hyperekplexia is missense and non-sense mutations in the glycine receptor (GlyR) alpha1 subunit gene (GLRA1) on chromosome 5q33.1, although we have also discovered rare mutations in the genes encoding the GlyR beta subunit (GLRB) and the GlyR clustering proteins gephyrin (GPNH) and collybistin (ARHGEF9). Recent studies of the Na(+)/Cl(-)-dependent glycine transporters GlyT1 and GlyT2 using mouse knockout models and human genetics have revealed that mutations in GlyT2 are a second major cause of hyperekplexia, while the phenotype of the GlyT1 knockout mouse resembles a devastating neurological disorder known as glycine encephalopathy (OMIM 605899). These findings highlight the importance of these transporters in regulating the levels of synaptic glycine.
Keywords: GlyT1; GlyT2; VIAAT; glycine encephalopathy; glycine transporters; hyperekplexia; startle disease.
Figures
Similar articles
-
The glycinergic system in human startle disease: a genetic screening approach.Front Mol Neurosci. 2010 Mar 23;3:8. doi: 10.3389/fnmol.2010.00008. eCollection 2010. Front Mol Neurosci. 2010. PMID: 20407582 Free PMC article.
-
Mutations in the gene encoding GlyT2 (SLC6A5) define a presynaptic component of human startle disease.Nat Genet. 2006 Jul;38(7):801-6. doi: 10.1038/ng1814. Epub 2006 Jun 4. Nat Genet. 2006. PMID: 16751771 Free PMC article.
-
Distinct phenotypes in zebrafish models of human startle disease.Neurobiol Dis. 2013 Dec;60:139-51. doi: 10.1016/j.nbd.2013.09.002. Epub 2013 Sep 9. Neurobiol Dis. 2013. PMID: 24029548 Free PMC article.
-
The genetics of hyperekplexia: more than startle!Trends Genet. 2008 Sep;24(9):439-47. doi: 10.1016/j.tig.2008.06.005. Epub 2008 Aug 15. Trends Genet. 2008. PMID: 18707791 Review.
-
Molecular mechanisms of glycine transporter GlyT2 mutations in startle disease.Biol Chem. 2012 Apr;393(4):283-9. doi: 10.1515/BC-2011-232. Biol Chem. 2012. PMID: 22114948 Review.
Cited by
-
A MusD retrotransposon insertion in the mouse Slc6a5 gene causes alterations in neuromuscular junction maturation and behavioral phenotypes.PLoS One. 2012;7(1):e30217. doi: 10.1371/journal.pone.0030217. Epub 2012 Jan 17. PLoS One. 2012. PMID: 22272310 Free PMC article.
-
Developmental seizures and mortality result from reducing GABAA receptor α2-subunit interaction with collybistin.Nat Commun. 2018 Aug 7;9(1):3130. doi: 10.1038/s41467-018-05481-1. Nat Commun. 2018. PMID: 30087324 Free PMC article.
-
Defects of the Glycinergic Synapse in Zebrafish.Front Mol Neurosci. 2016 Jun 29;9:50. doi: 10.3389/fnmol.2016.00050. eCollection 2016. Front Mol Neurosci. 2016. PMID: 27445686 Free PMC article. Review.
-
Frontiers in molecular neuroscience - résumé and perspective.Front Mol Neurosci. 2011 Dec 30;4:58. doi: 10.3389/fnmol.2011.00058. eCollection 2011. Front Mol Neurosci. 2011. PMID: 22232574 Free PMC article. No abstract available.
-
Can lesions to the motor cortex induce amyotrophic lateral sclerosis?J Neurol. 2014 Feb;261(2):283-90. doi: 10.1007/s00415-013-7185-7. Epub 2013 Nov 20. J Neurol. 2014. PMID: 24253481
References
Grants and funding
LinkOut - more resources
Full Text Sources