

A novel mutation in the sulfate transporter gene SLC26A2 (DTDST) specific to the Finnish population causes de la Chapelle dysplasia
- PMID: 18708426
- PMCID: PMC4361899
- DOI: 10.1136/jmg.2007.057158
A novel mutation in the sulfate transporter gene SLC26A2 (DTDST) specific to the Finnish population causes de la Chapelle dysplasia
Abstract
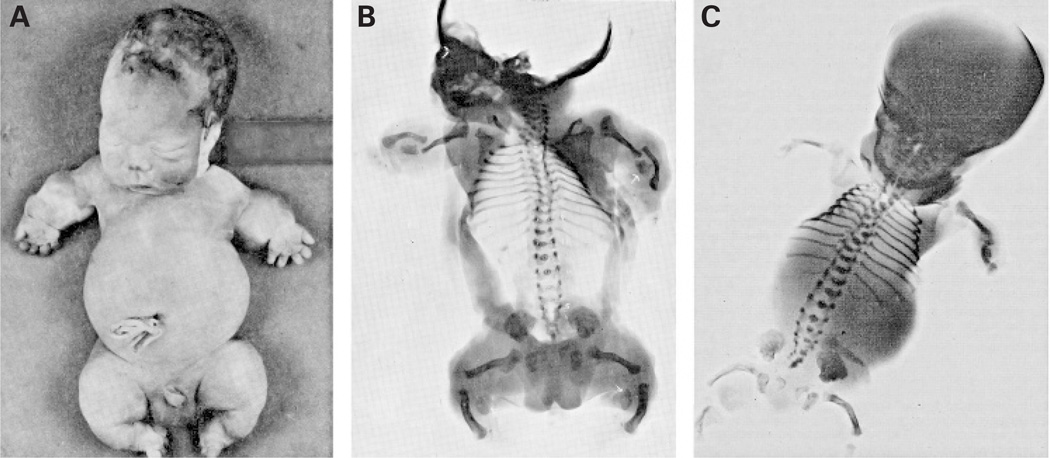
Background: Mutations in the sulfate transporter gene SLC26A2 (DTDST) cause a continuum of skeletal dysplasia phenotypes that includes achondrogenesis type 1B (ACG1B), atelosteogenesis type 2 (AO2), diastrophic dysplasia (DTD), and recessive multiple epiphyseal dysplasia (rMED). In 1972, de la Chapelle et al reported two siblings with a lethal skeletal dysplasia, which was denoted "neonatal osseous dysplasia" and "de la Chapelle dysplasia" (DLCD). It was suggested that DLCD might be part of the SLC26A2 spectrum of phenotypes, both because of the Finnish origin of the original family and of radiographic similarities to ACG1B and AO2.
Objective: To test the hypothesis whether SLC26A2 mutations are responsible for DLCD.
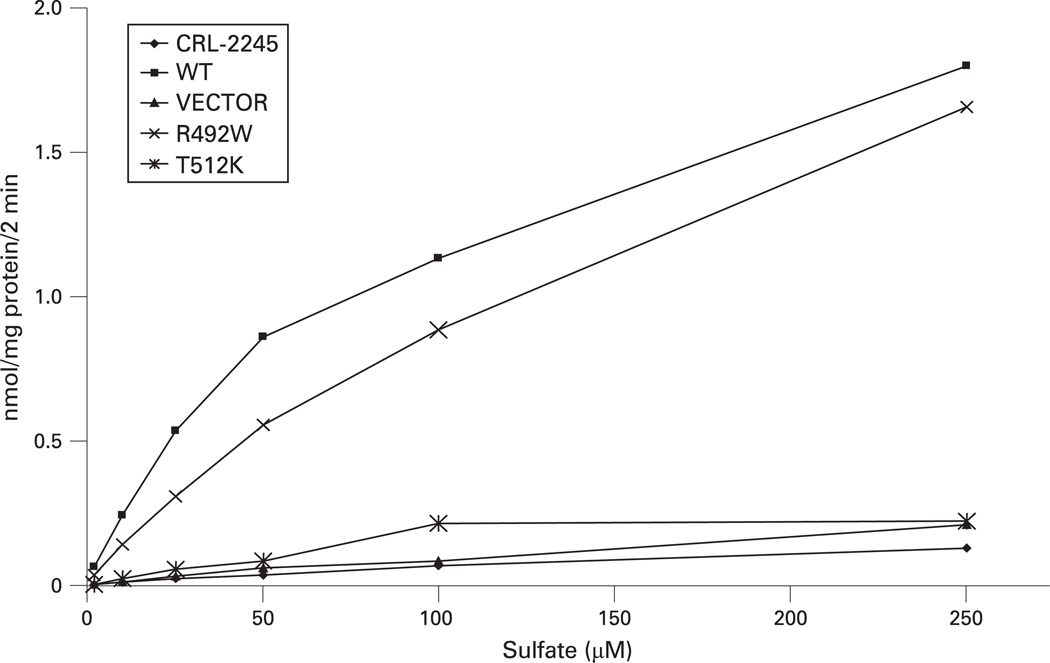
Methods: We studied the DNA from the original DLCD family and from seven Finnish DTD patients in whom we had identified only one copy of IVS1+2T>C, the common Finnish mutation. A novel SLC26A2 mutation was found in all subjects, inserted by site-directed mutagenesis in a vector harbouring the SLC26A2 cDNA, and expressed in sulfate transport deficient Chinese hamster ovary (CHO) cells to measure sulfate uptake activity.
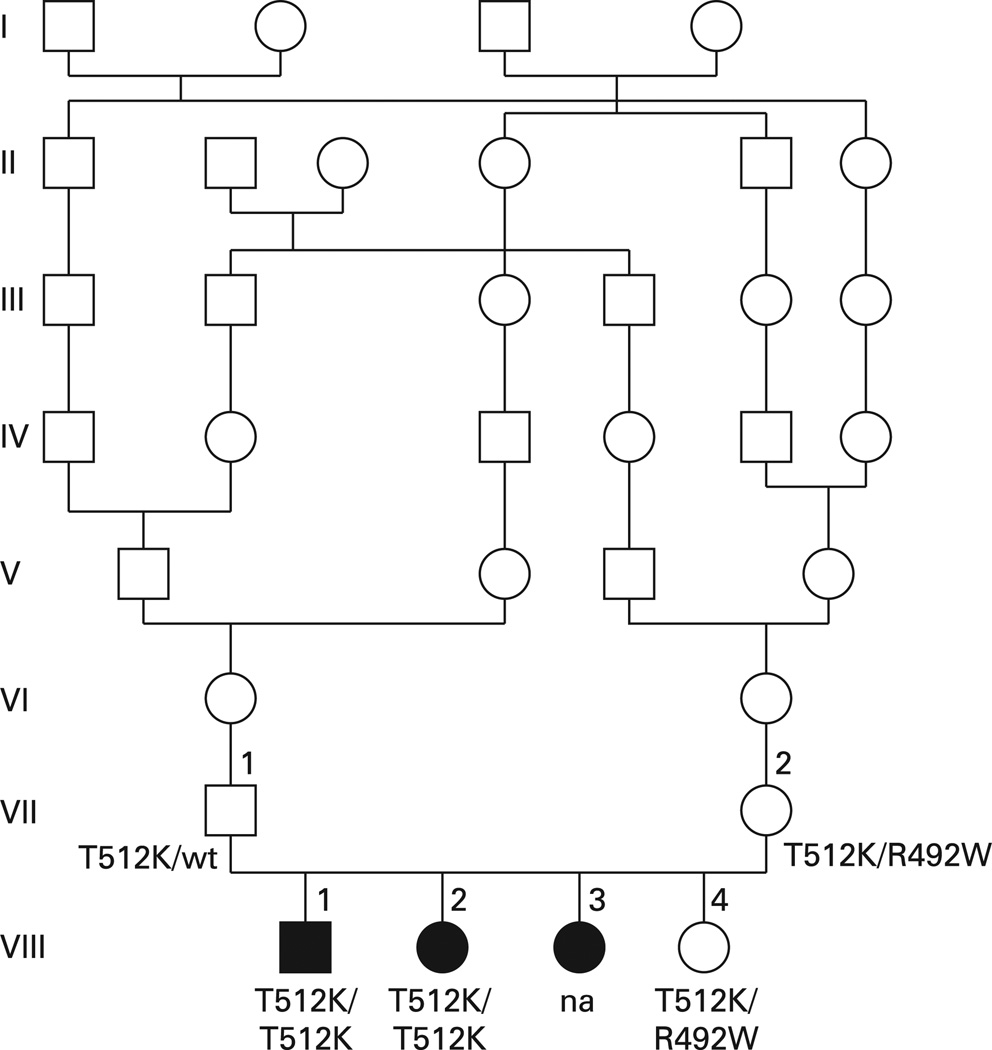
Results: We identified a hitherto undescribed SLC26A2 mutation, T512K, homozygous in the affected subjects and heterozygous in both parents and in the unaffected sister. T512K was then identified as second pathogenic allele in the seven Finnish DTD subjects. Expression studies confirmed pathogenicity.
Conclusions: DLCD is indeed allelic to the other SLC26A2 disorders. T512K is a second rare "Finnish" mutation that results in DLCD at homozygosity and in DTD when compounded with the milder, common Finnish mutation.
Figures
Similar articles
-
Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance.Hum Mutat. 2001 Mar;17(3):159-71. doi: 10.1002/humu.1. Hum Mutat. 2001. PMID: 11241838
-
Genotype-phenotype correlation in DTDST dysplasias: Atelosteogenesis type II and diastrophic dysplasia variant in one family.Am J Med Genet A. 2010 Dec;152A(12):3043-50. doi: 10.1002/ajmg.a.33736. Am J Med Genet A. 2010. PMID: 21077202 Review.
-
SLC26A2 disease spectrum in Sweden - high frequency of recessive multiple epiphyseal dysplasia (rMED).Clin Genet. 2015 Mar;87(3):273-8. doi: 10.1111/cge.12371. Epub 2014 Apr 1. Clin Genet. 2015. PMID: 24598000
-
A compound heterozygote SLC26A2 mutation resulting in robin sequence, mild limbs shortness, accelerated carpal ossification, and multiple epiphysial dysplasia in two Brazilian sisters. A new intermediate phenotype between diastrophic dysplasia and recessive multiple epiphyseal dysplasia.Am J Med Genet A. 2013 Aug;161A(8):2088-94. doi: 10.1002/ajmg.a.36057. Epub 2013 Jul 9. Am J Med Genet A. 2013. PMID: 23840040
-
SLC26A2-Associated Diastrophic Dysplasia and rMED-Clinical Features in Affected Finnish Children and Review of the Literature.Genes (Basel). 2021 May 11;12(5):714. doi: 10.3390/genes12050714. Genes (Basel). 2021. PMID: 34064542 Free PMC article. Review.
Cited by
-
SLC26A2/DTDST Spectrum: A Cohort of 12 Patients Associated with a Comprehensive Review of the Genotype-Phenotype Correlation.Mol Syndromol. 2023 Jan;13(6):485-495. doi: 10.1159/000525020. Epub 2022 Jun 15. Mol Syndromol. 2023. PMID: 36660027 Free PMC article.
-
Skeletal Dysplasias Caused by Sulfation Defects.Int J Mol Sci. 2020 Apr 14;21(8):2710. doi: 10.3390/ijms21082710. Int J Mol Sci. 2020. PMID: 32295296 Free PMC article. Review.
-
Recessive multiple epiphyseal dysplasia and Stargardt disease in two sisters.Mol Genet Genomic Med. 2021 Apr;9(4):e1630. doi: 10.1002/mgg3.1630. Epub 2021 Mar 16. Mol Genet Genomic Med. 2021. PMID: 33724725 Free PMC article.
-
Biallelic variants in SLC26A2 cause multiple epiphyseal dysplasia-4 by disturbing chondrocyte homeostasis.Orphanet J Rare Dis. 2024 Jul 2;19(1):245. doi: 10.1186/s13023-024-03228-4. Orphanet J Rare Dis. 2024. PMID: 38956600 Free PMC article.
-
Altered responsiveness to TGF-β results in reduced Papss2 expression and alterations in the biomechanical properties of mouse articular cartilage.Arthritis Res Ther. 2012 Mar 6;14(2):R49. doi: 10.1186/ar3762. Arthritis Res Ther. 2012. PMID: 22394585 Free PMC article.
References
-
- De la Chapelle A, Maroteaux P, Havu N, Granroth G. A rare lethal bone dysplasia with recessive autosomic transmission. Arch Fr Pediatr. 1972;29:759–770. - PubMed
-
- Whitley CB, Burke BA, Granroth G, Gorlin RJ. de la Chapelle dysplasia. Am J Med Genet. 1986;25:29–39. - PubMed
-
- Sillence DO, Kozlowski K, Rogers JG, Sprague PL, Cullity GJ, Osborn RA. Atelosteogenesis: evidence for heterogeneity. Pediatr Radiol. 1987;17:112–118. - PubMed
-
- Schrander-Stumpel C, Havenith M, Linden EV, Maertzdorf W, Offermans J, van der Harten J. De la Chapelle dysplasia (atelosteogenesis type II): case report and review of the literature [corrected] Clin Dysmorphol. 1994;3:318–327. - PubMed
-
- Rossi A, van der Harten HJ, Beemer FA, Kleijer WJ, Gitzelmann R, Steinmann B, Superti-Furga A. Phenotypic and genotypic overlap between atelosteogenesis type 2 and diastrophic dysplasia. Hum Genet. 1996;98:657–661. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources