

Review
doi: 10.1016/j.bone.2008.05.013.
Epub 2008 May 28.
Insights from a rare genetic disorder of extra-skeletal bone formation, fibrodysplasia ossificans progressiva (FOP)
Affiliations
- PMID: 18590993
- PMCID: PMC2601573
- DOI: 10.1016/j.bone.2008.05.013
Item in Clipboard
Review
Insights from a rare genetic disorder of extra-skeletal bone formation, fibrodysplasia ossificans progressiva (FOP)
Bone.
2008 Sep.
Abstract
Fibrodysplasia ossificans progressiva (FOP) is a rare human genetic disorder of extensive and debilitating extra-skeletal bone formation. While the challenges of investigating a rare condition are many, the potential benefits are also great - not only for the specific disease under investigation, but also for the unique perspective on how cells normally function and the mechanisms that underlie more common disorders. This review will illustrate some of the many insights that we have gained by studying FOP.
Figures
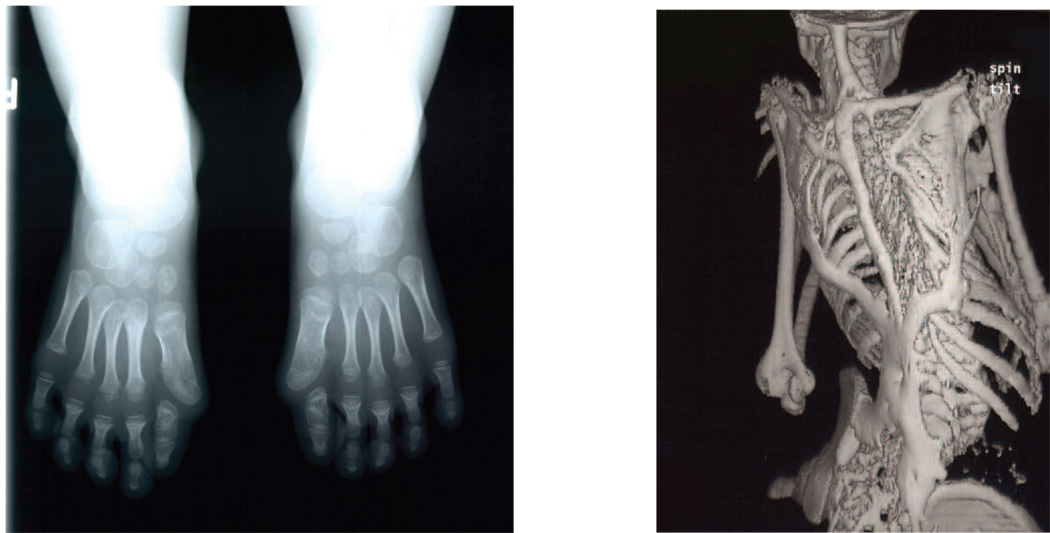
Classic fibrodysplasia ossificans progressiva (FOP) is defined by two characteristic clinical features. (a) Extensive heterotopic bone formation is seen in this 3-dimensional reconstructed computed tomography (CT) scan of the back of a twelve-year-old child. Flareups of FOP arise and progress in a well-defined spatial pattern that result in ribbons, sheets, and plates of bone that fuse the joints of the axial and appendicular skeleton, entombing the patient in a “second skeleton” of heterotopic bone. (b) An anteroposterior radiograph of the feet of a three-year-old child shows symmetrical great toe malformations of metatarsals and proximal phalanges along with microdactyly, fused interphalangeal joints, and hallux valgus deviations at the metatarsophalangeal joints.
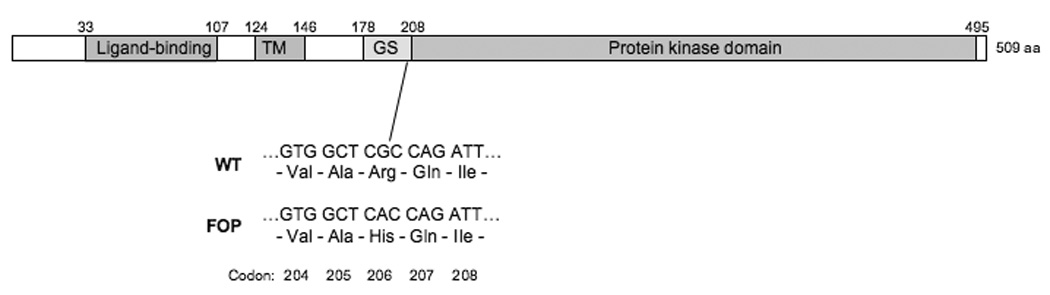
A schematic of the ACVR1 protein is shown, with key functional domains indicated: the extracellular BMP ligand-binding domain, the single trans-membrane (TM) domain, the glycine-serine (GS) activation domain, and the protein kinase signaling domain. Amino acid positions of each domain boundary are indicated above the schematic. All patients who show the classic characteristic features of FOP have the identical mutation in codon 206 within the GS domain. The nucleotide and amino acid sequences of codons 204–208 are shown for the wild-type and FOP mutation. The classic FOP mutation (a G to A substitution at nucleotide position 617 of the cDNA) causes a substitution of histidine for arginine (Arg206His).
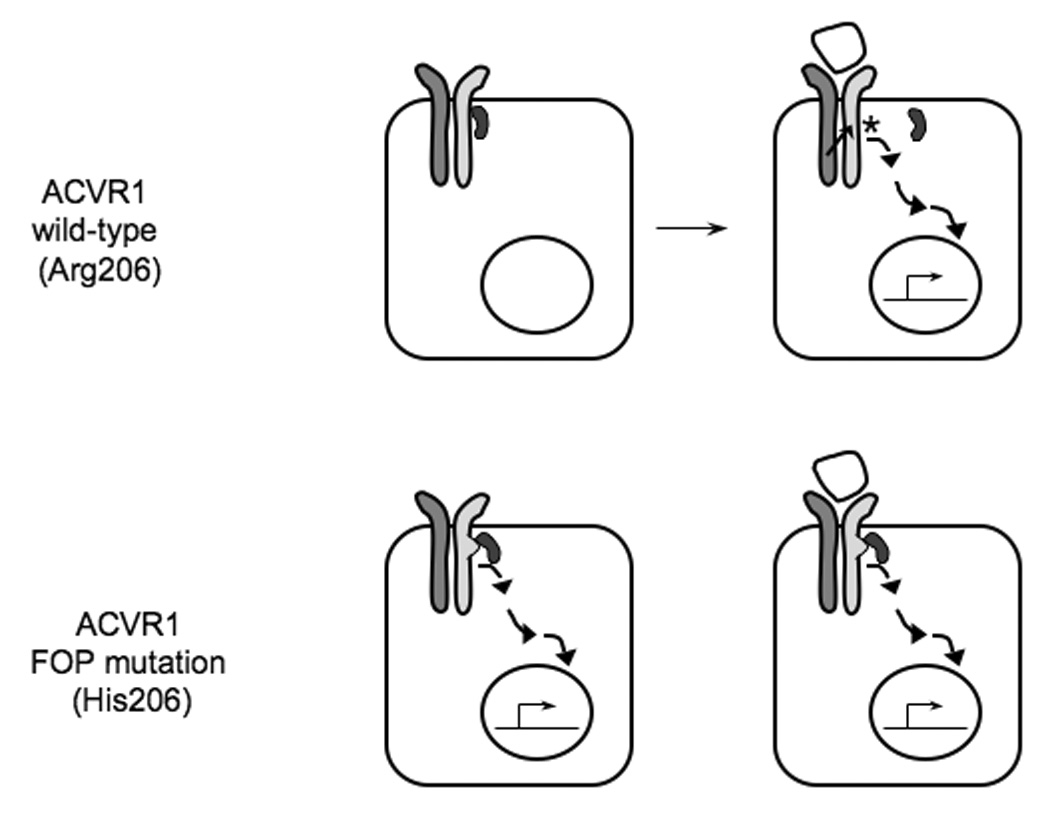
In the absence of BMP ligand, FKBP12 binds the wild-type type I BMP receptor (with arginine at codon 206; Arg206) and prevents “leaky” activation of downstream signaling in the absence of ligand. Upon BMP binding to the type I/type II BMP receptor complex, the type II receptor phosphorylates (asterisk) the type I receptor within the GS domain and induces the release of FKBP12 from the receptor and activation of downstream signaling. We hypothesize that FKBP12 does not correctly bind to the mutant FOP ACVR1 type I receptor (with histidine at codon 206; His206) either in the presence or the absence of BMP-receptor binding, allowing increased activation of BMP signaling.
Similar articles
-
Fibrodysplasia ossificans progressiva: a human genetic disorder of extraskeletal bone formation, or--how does one tissue become another?Wiley Interdiscip Rev Dev Biol. 2012 Jan-Feb;1(1):153-65. doi: 10.1002/wdev.9. Wiley Interdiscip Rev Dev Biol. 2012. PMID: 22408652 Free PMC article.
-
Palovarotene Inhibits Heterotopic Ossification and Maintains Limb Mobility and Growth in Mice With the Human ACVR1(R206H) Fibrodysplasia Ossificans Progressiva (FOP) Mutation.J Bone Miner Res. 2016 Sep;31(9):1666-75. doi: 10.1002/jbmr.2820. Epub 2016 Mar 12. J Bone Miner Res. 2016. PMID: 26896819 Free PMC article.
-
Heterotopic Ossification in Mouse Models of Fibrodysplasia Ossificans Progressiva.Methods Mol Biol. 2019;1891:247-255. doi: 10.1007/978-1-4939-8904-1_18. Methods Mol Biol. 2019. PMID: 30414138 Free PMC article.
-
Intersections of Fibrodysplasia Ossificans Progressiva and Traumatic Heterotopic Ossification.Biomolecules. 2024 Mar 14;14(3):349. doi: 10.3390/biom14030349. Biomolecules. 2024. PMID: 38540768 Free PMC article. Review.
-
Fibrodysplasia Ossificans Progressiva: What Have We Achieved and Where Are We Now? Follow-up to the 2015 Lorentz Workshop.Front Endocrinol (Lausanne). 2021 Nov 10;12:732728. doi: 10.3389/fendo.2021.732728. eCollection 2021. Front Endocrinol (Lausanne). 2021. PMID: 34858325 Free PMC article. Review.
Cited by
-
The role of bone morphogenetic proteins in ankylosing spondylitis.Ther Adv Musculoskelet Dis. 2012 Aug;4(4):293-9. doi: 10.1177/1759720X12444175. Ther Adv Musculoskelet Dis. 2012. PMID: 22859928 Free PMC article.
-
Overactive bone morphogenetic protein signaling in heterotopic ossification and Duchenne muscular dystrophy.Cell Mol Life Sci. 2013 Feb;70(3):407-23. doi: 10.1007/s00018-012-1054-x. Epub 2012 Jul 4. Cell Mol Life Sci. 2013. PMID: 22752156 Free PMC article. Review.
-
Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype.Semin Cancer Biol. 2012 Oct;22(5-6):379-84. doi: 10.1016/j.semcancer.2012.04.004. Epub 2012 Apr 23. Semin Cancer Biol. 2012. PMID: 22554794 Free PMC article. Review.
-
Fibrodysplasia ossificans progressiva (FOP): watch the great toes!Eur J Pediatr. 2010 Nov;169(11):1417-21. doi: 10.1007/s00431-010-1232-5. Epub 2010 Jun 26. Eur J Pediatr. 2010. PMID: 20577760 Free PMC article.
-
Bone morphogenetic protein receptor signal transduction in human disease.J Pathol. 2019 Jan;247(1):9-20. doi: 10.1002/path.5170. Epub 2018 Nov 27. J Pathol. 2019. PMID: 30246251 Free PMC article. Review.
References
-
- Harvey W. Letters. In: Alfred Fishman M, editor. The Works of William Harvey. Philadelphia: University of Pennsylvania Press; 1989. pp. 616–617.
-
- Kruglyak The road to genome-wide association studies. Nature Reviews Genetics. 2008;9 - PubMed
-
- Shore EM, Feldman GJ, Xu M, Kaplan FS. The genetics of fibrodysplasia ossificans progressiva. Clinical Reviews in Bone and Mineral Metabolism. 2005;3:201–204.
-
- McKusick VA. Heritable Disorders of Connective Tissue. 4th ed. St. Louis, MO: C.V. Mosby; 1972.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
