

Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4)
- PMID: 17503324
- PMCID: PMC1867094
- DOI: 10.1086/518314
Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4)
Erratum in
- Am J Hum Genet. 2007 Sep;81(3):634
Abstract
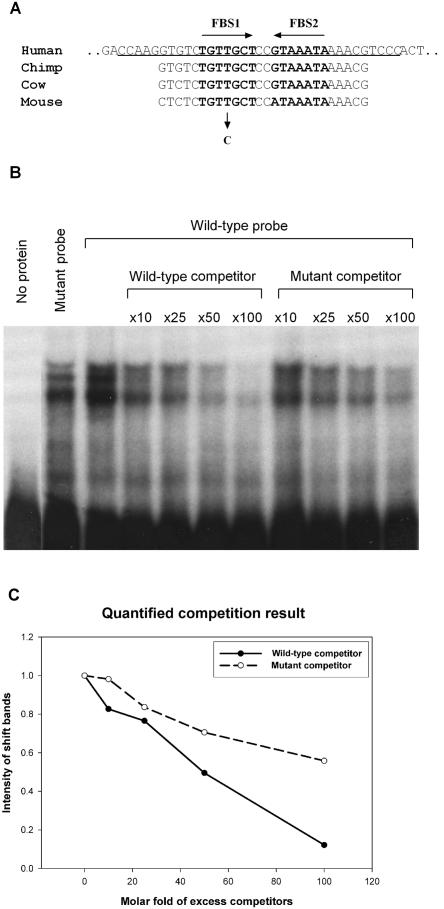
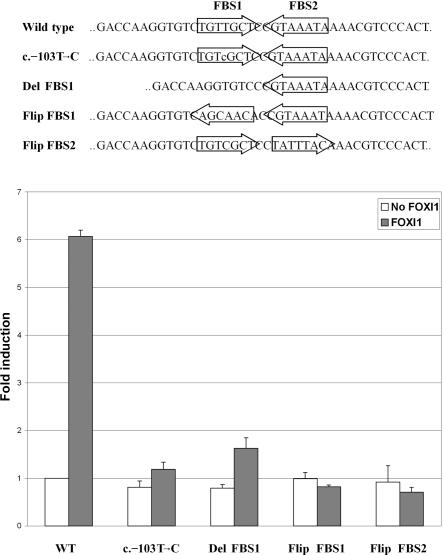
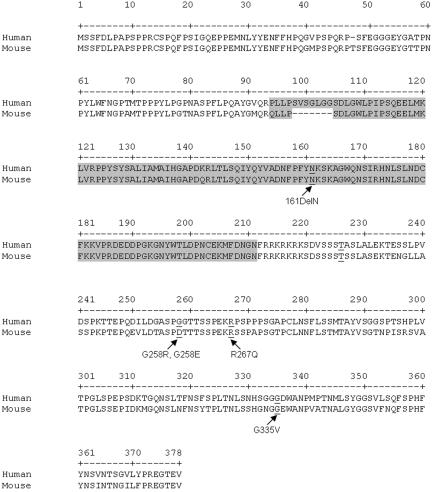
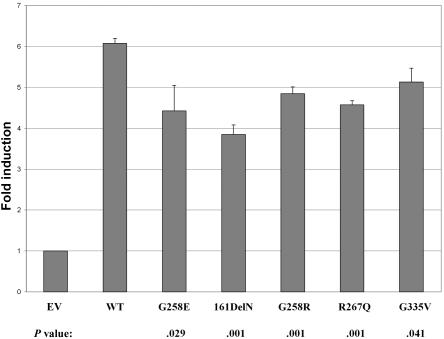
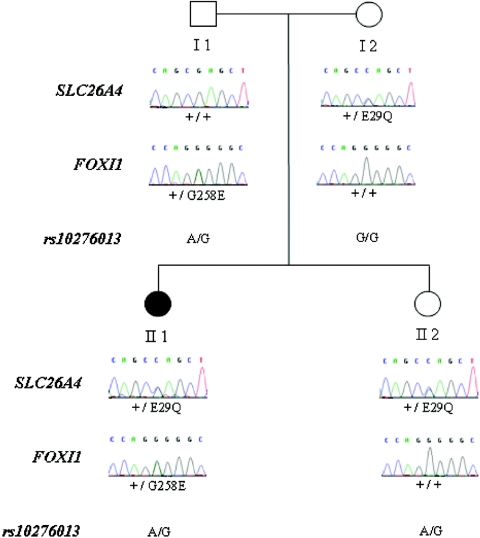
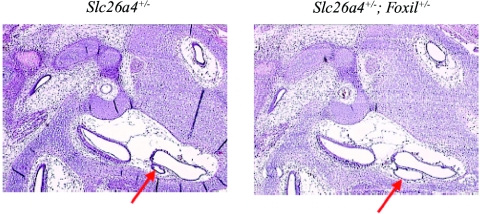
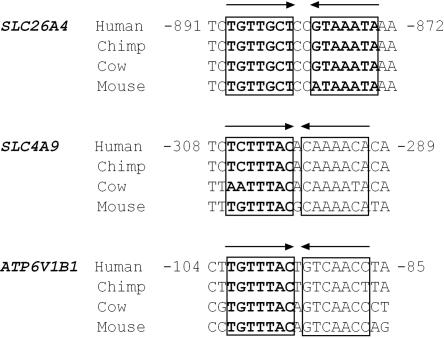
Although recessive mutations in the anion transporter gene SLC26A4 are known to be responsible for Pendred syndrome (PS) and nonsyndromic hearing loss associated with enlarged vestibular aqueduct (EVA), also known as "DFNB4," a large percentage of patients with this phenotype lack mutations in the SLC26A4 coding region in one or both alleles. We have identified and characterized a key transcriptional regulatory element in the SLC26A4 promoter that binds FOXI1, a transcriptional activator of SLC26A4. In nine patients with PS or nonsyndromic EVA, a novel c.-103T-->C mutation in this regulatory element interferes with FOXI1 binding and completely abolishes FOXI1-mediated transcriptional activation. We have also identified six patients with mutations in FOXI1 that compromise its ability to activate SLC26A4 transcription. In one family, the EVA phenotype segregates in a double-heterozygous mode in the affected individual who carries single mutations in both SLC26A4 and FOXI1. This finding is consistent with our observation that EVA occurs in the Slc26a4(+/-); Foxi1(+/-) double-heterozygous mouse mutant. These results support a novel dosage-dependent model for the molecular pathogenesis of PS and nonsyndromic EVA that involves SLC26A4 and its transcriptional regulatory machinery.
Figures
Similar articles
-
Phenotypic analyses and mutation screening of the SLC26A4 and FOXI1 genes in 101 Taiwanese families with bilateral nonsyndromic enlarged vestibular aqueduct (DFNB4) or Pendred syndrome.Audiol Neurootol. 2010;15(1):57-66. doi: 10.1159/000231567. Epub 2009 Aug 1. Audiol Neurootol. 2010. PMID: 19648736
-
Analysis of SLC26A4, FOXI1, and KCNJ10 Gene Variants in Patients with Incomplete Partition of the Cochlea and Enlarged Vestibular Aqueduct (EVA) Anomalies.Int J Mol Sci. 2022 Dec 6;23(23):15372. doi: 10.3390/ijms232315372. Int J Mol Sci. 2022. PMID: 36499699 Free PMC article.
-
Analysis of the thyroid phenotype in 42 patients with Pendred syndrome and nonsyndromic enlargement of the vestibular aqueduct.Thyroid. 2014 Apr;24(4):639-48. doi: 10.1089/thy.2013.0164. Epub 2014 Jan 20. Thyroid. 2014. PMID: 24224479
-
Genetic architecture and phenotypic landscape of SLC26A4-related hearing loss.Hum Genet. 2022 Apr;141(3-4):455-464. doi: 10.1007/s00439-021-02311-1. Epub 2021 Aug 3. Hum Genet. 2022. PMID: 34345941 Review.
-
SLC26A4 genotypes and phenotypes associated with enlargement of the vestibular aqueduct.Cell Physiol Biochem. 2011;28(3):545-52. doi: 10.1159/000335119. Epub 2011 Nov 18. Cell Physiol Biochem. 2011. PMID: 22116369 Free PMC article. Review.
Cited by
-
The role of foxi family transcription factors in the development of the ear and jaw.Curr Top Dev Biol. 2015;111:461-95. doi: 10.1016/bs.ctdb.2014.11.014. Epub 2015 Jan 21. Curr Top Dev Biol. 2015. PMID: 25662269 Free PMC article. Review.
-
Etiology and audiological outcomes at 3 years for 364 children in Australia.PLoS One. 2013;8(3):e59624. doi: 10.1371/journal.pone.0059624. Epub 2013 Mar 28. PLoS One. 2013. PMID: 23555729 Free PMC article.
-
Unresolved questions regarding human hereditary deafness.Oral Dis. 2017 Jul;23(5):551-558. doi: 10.1111/odi.12516. Epub 2016 Jul 11. Oral Dis. 2017. PMID: 27259978 Free PMC article. Review.
-
Foxi1 inactivation rescues loss of principal cell fate selection in Hes1-deficient kidneys but does not ensure maintenance of principal cell gene expression.Dev Biol. 2020 Oct 1;466(1-2):1-11. doi: 10.1016/j.ydbio.2020.08.005. Epub 2020 Aug 13. Dev Biol. 2020. PMID: 32800756 Free PMC article.
-
Mechanism of anion exchange and small-molecule inhibition of pendrin.Nat Commun. 2024 Jan 6;15(1):346. doi: 10.1038/s41467-023-44612-1. Nat Commun. 2024. PMID: 38184688 Free PMC article.
References
Web Resources
-
- GenBank, http://ncbi.nlm.nih.gov/Genbank/ (for SLC26A4 [accession number NC_000007.12], Slc26a4 [accession number NC_000078.4], Foxi1 [accession number NP_076396.2], FOXI1 [accession number NP_036320.2], Slc4a9 [accession number NC_000084.4], Atp6v1b1 [accession number NC_000072.4], SLC4A9 [accession number NC_000005.8], and ATP6V1B1 [accession number NC_000002.10])
-
- Hereditary Hearing Loss Homepage, http://webh01.ua.ac.be/hhh/
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.gov/Omim/ (for PS, SLC26A4, EVA, DFNB4, FOXI1, POU3F4, GJB2, Slc4a9, Atp6v1b1, FOXP2, EYA1, SIX1, BOR, EYA4, DFNA10, POU4F3, DFNA15, DFN3, GRHL2, DFNA28, PAX3, MITF, SNAI2, SOX10, WS1, WS2A, WS3, and WS4)
-
- UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway/ (for the genomic sequence alignment)
References
-
- Batsakis JG, Nishiyama RH (1962) Deafness with sporadic goiter: Pendred’s syndrome. Arch Otolaryngol 76:401–406 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
