

Laing early onset distal myopathy: slow myosin defect with variable abnormalities on muscle biopsy
- PMID: 16103042
- PMCID: PMC2077563
- DOI: 10.1136/jnnp.2005.073825
Laing early onset distal myopathy: slow myosin defect with variable abnormalities on muscle biopsy
Abstract
Background: Laing early onset distal myopathy (MPD1) is an autosomal dominant myopathy caused by mutations within the slow skeletal muscle fibre myosin heavy chain gene, MYH7. It is allelic with myosin storage myopathy, with the commonest form of familial hypertrophic cardiomyopathy, and with one form of dilated cardiomyopathy. However, the clinical picture of MPD1 is distinct from these three conditions.
Objective: To collate and discuss the histological features reported in the muscle biopsies of MPD1 patients and to outline the clinical features.
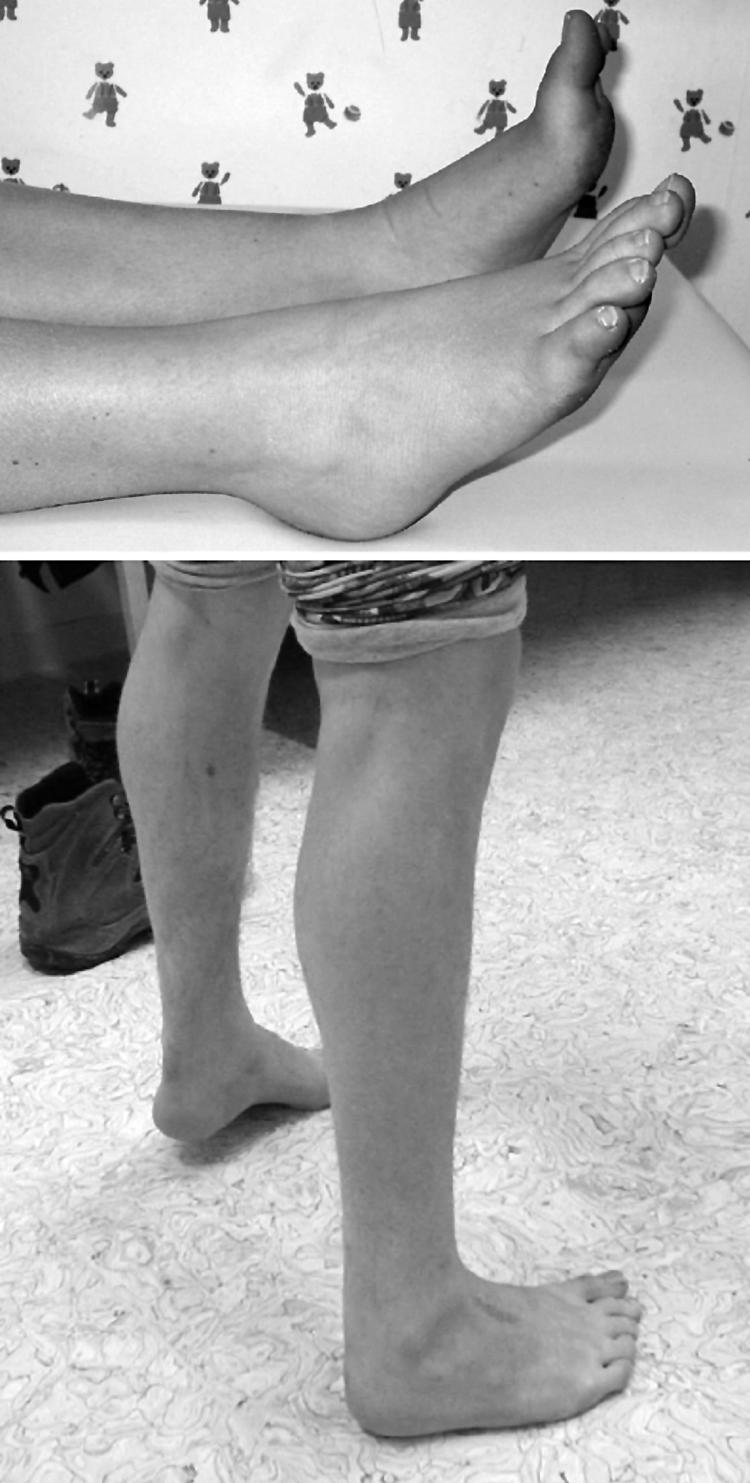
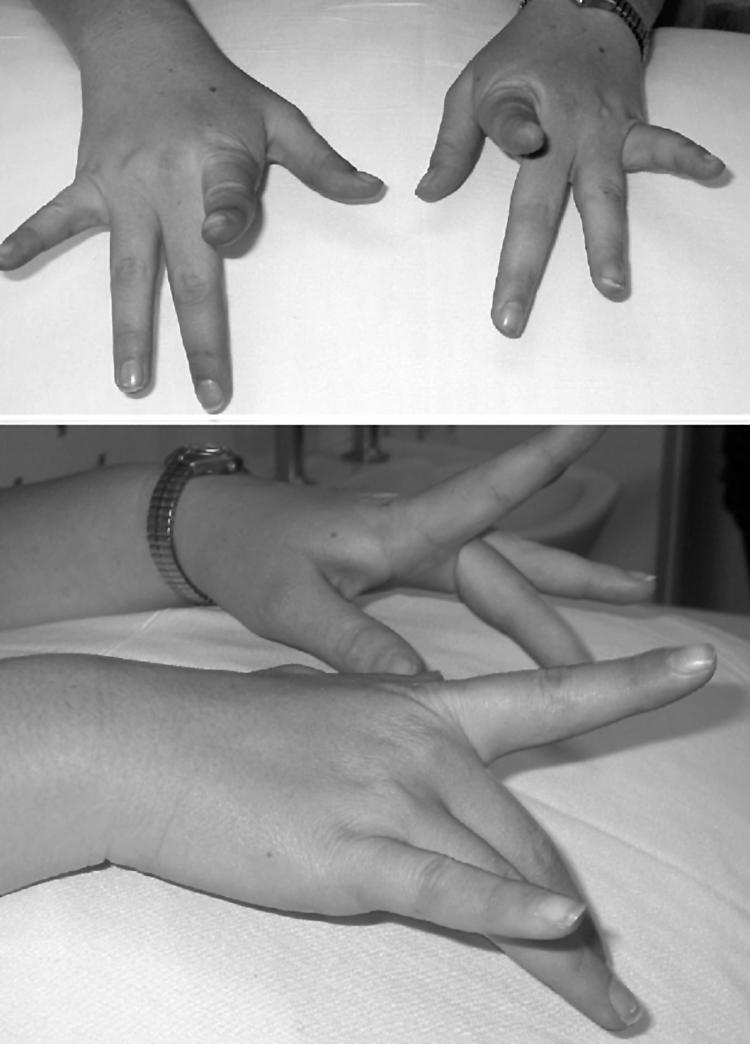
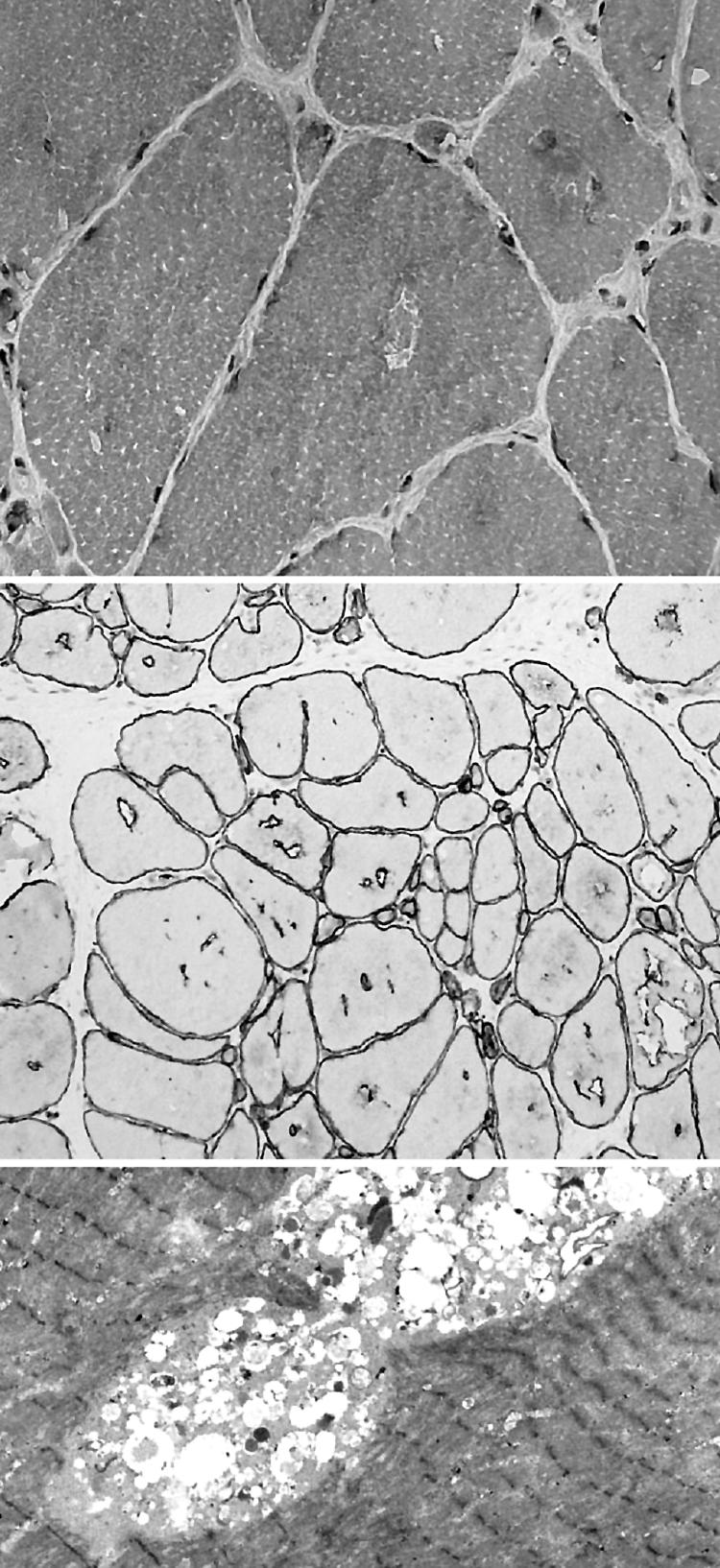
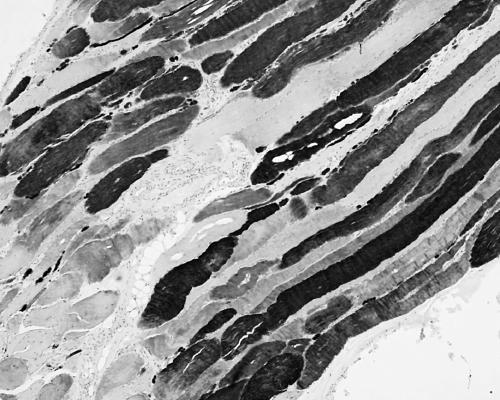
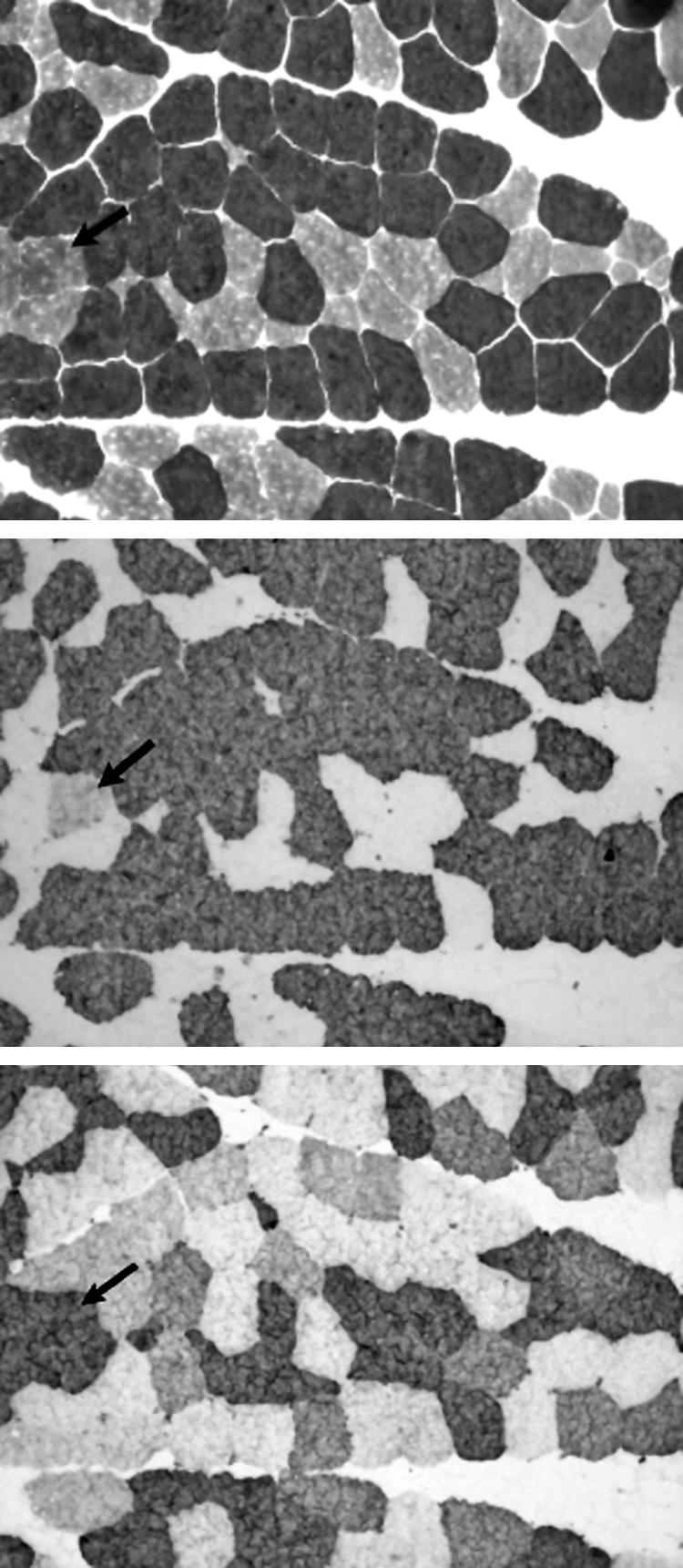
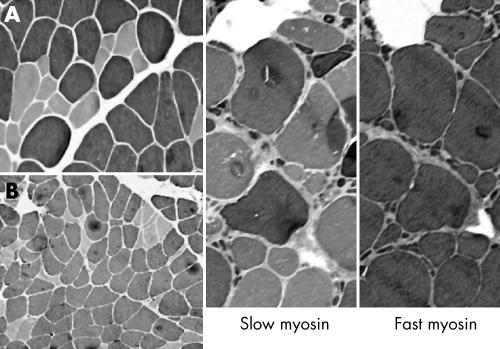
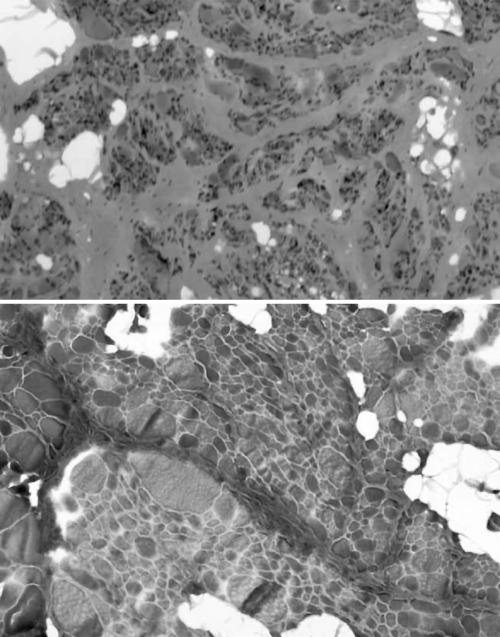
Results: The phenotype of MPD1 was consistent, with initial weakness of great toe/ankle dorsiflexion, and later development of weakness of finger extension and neck flexion. Age of onset was the only variable, being from birth up to the 20 s, but progression was always very slow. The pathological features were variable. In this retrospective series, there were no pathognomonic diagnostic features, although atrophic type I fibres were found in half the families. Rimmed vacuoles are consistently seen in all other distal myopathies with the exception of Myoshi distal myopathy. However, they were found in a minority of patients with MPD1, and were not prominent when present. Immunohistochemical staining for slow and fast myosin showed co-expression of slow and fast myosin in some type I fibres, possibly indicating a switch to type II status. This may be a useful aid to diagnosis.
Conclusions: The pathological findings in MPD1 are variable and appear to be affected by factors such as the specific muscle biopsied, the age of the patient at biopsy, and the duration of disease manifestations.
Conflict of interest statement
Competing interests: none declared
Similar articles
-
Novel mutations widen the phenotypic spectrum of slow skeletal/β-cardiac myosin (MYH7) distal myopathy.Hum Mutat. 2014 Jul;35(7):868-79. doi: 10.1002/humu.22553. Epub 2014 May 21. Hum Mutat. 2014. PMID: 24664454 Free PMC article.
-
Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause laing early-onset distal myopathy (MPD1).Am J Hum Genet. 2004 Oct;75(4):703-8. doi: 10.1086/424760. Epub 2004 Aug 20. Am J Hum Genet. 2004. PMID: 15322983 Free PMC article.
-
Complex sarcolemmal invaginations mimicking myotendinous junctions in a case of Laing early-onset distal myopathy.Neuropathology. 2015 Dec;35(6):575-81. doi: 10.1111/neup.12220. Epub 2015 Jun 10. Neuropathology. 2015. PMID: 26094647
-
Hereditary myosin myopathies.Neuromuscul Disord. 2007 May;17(5):355-67. doi: 10.1016/j.nmd.2007.02.008. Epub 2007 Apr 16. Neuromuscul Disord. 2007. PMID: 17434305 Review.
-
Thick filament diseases.Adv Exp Med Biol. 2008;642:78-91. doi: 10.1007/978-0-387-84847-1_7. Adv Exp Med Biol. 2008. PMID: 19181095 Review.
Cited by
-
Novel mutations widen the phenotypic spectrum of slow skeletal/β-cardiac myosin (MYH7) distal myopathy.Hum Mutat. 2014 Jul;35(7):868-79. doi: 10.1002/humu.22553. Epub 2014 May 21. Hum Mutat. 2014. PMID: 24664454 Free PMC article.
-
A Laing distal myopathy-associated proline substitution in the β-myosin rod perturbs myosin cross-bridging activity.J Clin Invest. 2024 May 1;134(9):e172599. doi: 10.1172/JCI172599. J Clin Invest. 2024. PMID: 38690726 Free PMC article.
-
Laing distal myopathy pathologically resembling inclusion body myositis.Ann Clin Transl Neurol. 2014 Dec;1(12):1053-8. doi: 10.1002/acn3.140. Epub 2014 Nov 6. Ann Clin Transl Neurol. 2014. PMID: 25574480 Free PMC article.
-
Sarcomeric myopathies associated with tremor: new insights and perspectives.J Muscle Res Cell Motil. 2020 Dec;41(4):285-295. doi: 10.1007/s10974-019-09559-1. Epub 2019 Oct 16. J Muscle Res Cell Motil. 2020. PMID: 31620961 Free PMC article.
-
Novel mutations in MYBPC1 are associated with myogenic tremor and mild myopathy.Ann Neurol. 2019 Jul;86(1):129-142. doi: 10.1002/ana.25494. Epub 2019 May 17. Ann Neurol. 2019. PMID: 31025394 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical