

Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations
- PMID: 15863612
- PMCID: PMC1149428
- DOI: 10.1073/pnas.0502506102
Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations
Abstract
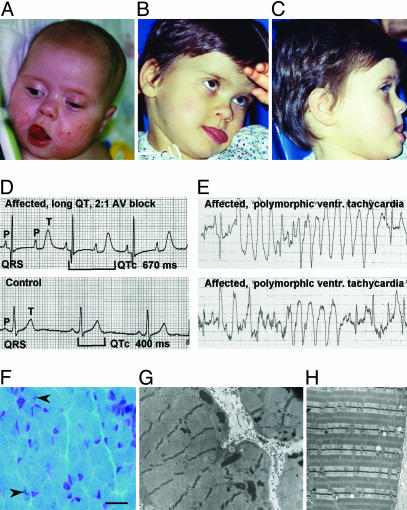
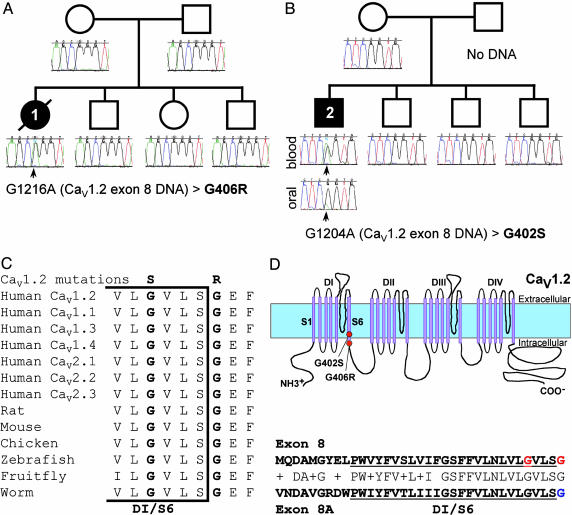
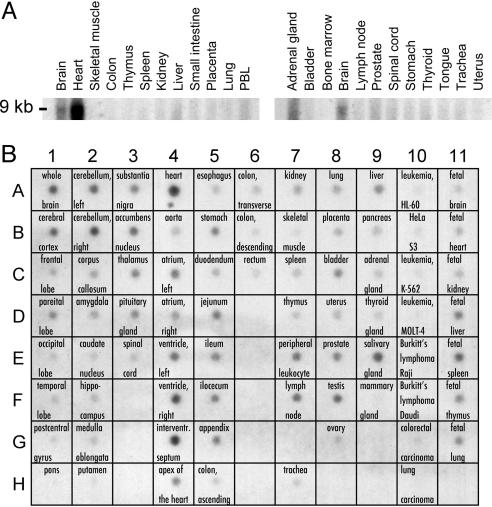
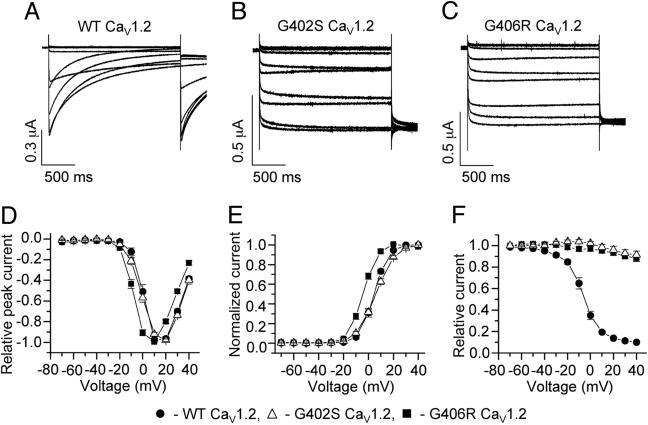
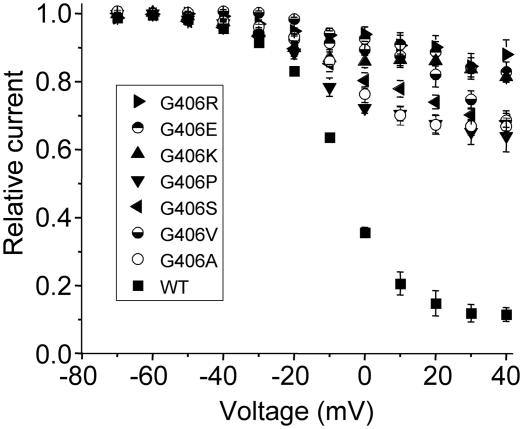
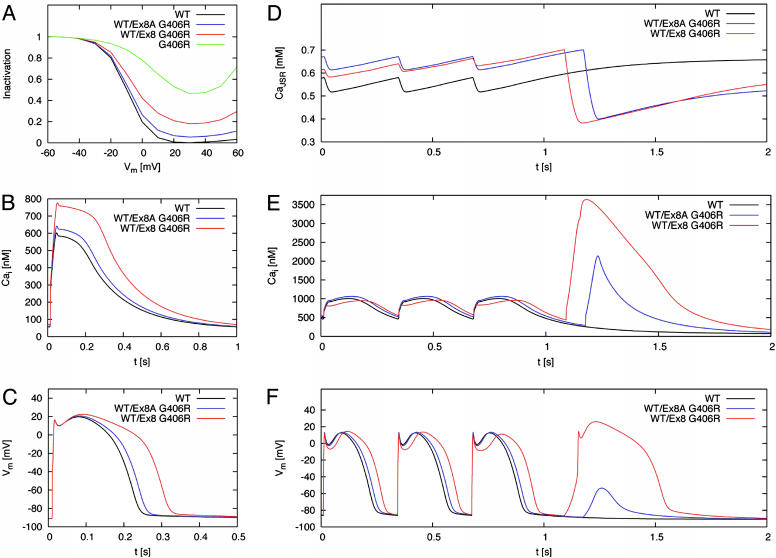
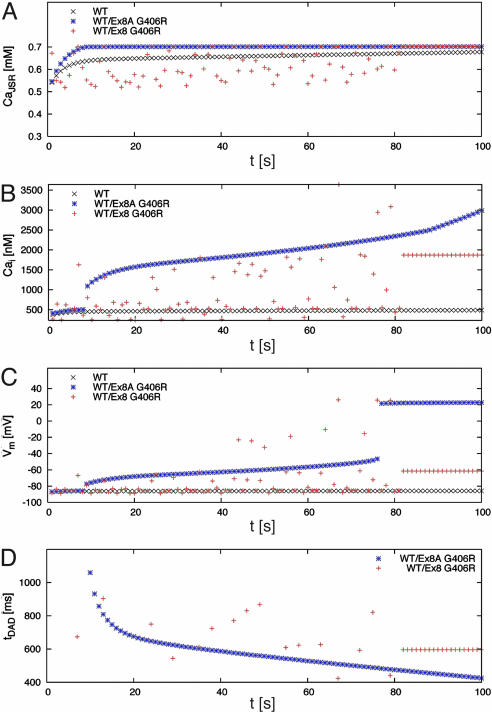
Timothy syndrome (TS) is a multisystem disorder that causes syncope and sudden death from cardiac arrhythmias. Prominent features include congenital heart disease, immune deficiency, intermittent hypoglycemia, cognitive abnormalities, and autism. All TS individuals have syndactyly (webbing of fingers and toes). We discovered that TS resulted from a recurrent, de novo cardiac L-type calcium channel (CaV1.2) mutation, G406R. G406 is located in alternatively spliced exon 8A, encoding transmembrane segment S6 of domain I. Here, we describe two individuals with a severe variant of TS (TS2). Neither child had syndactyly. Both individuals had extreme prolongation of the QT interval on electrocardiogram, with a QT interval corrected for heart rate ranging from 620 to 730 ms, causing multiple arrhythmias and sudden death. One individual had severe mental retardation and nemaline rod skeletal myopathy. We identified de novo missense mutations in exon 8 of CaV1.2 in both individuals. One was an analogous mutation to that found in exon 8A in classic TS, G406R. The other mutation was G402S. Exon 8 encodes the same region as exon 8A, and the two are mutually exclusive. The spliced form of CaV1.2 containing exon 8 is highly expressed in heart and brain, accounting for approximately 80% of CaV1.2 mRNAs. G406R and G402S cause reduced channel inactivation, resulting in maintained depolarizing L-type calcium currents. Computer modeling showed prolongation of cardiomyocyte action potentials and delayed afterdepolarizations, factors that increase risk of arrhythmia. These data indicate that gain-of-function mutations of CaV1.2 exons 8 and 8A cause distinct forms of TS.
Figures
Similar articles
-
Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism.Cell. 2004 Oct 1;119(1):19-31. doi: 10.1016/j.cell.2004.09.011. Cell. 2004. PMID: 15454078
-
Impaired chromaffin cell excitability and exocytosis in autistic Timothy syndrome TS2-neo mouse rescued by L-type calcium channel blockers.J Physiol. 2019 Mar;597(6):1705-1733. doi: 10.1113/JP277487. Epub 2019 Jan 28. J Physiol. 2019. PMID: 30629744 Free PMC article.
-
Gain-of-function mutations in the calcium channel CACNA1C (Cav1.2) cause non-syndromic long-QT but not Timothy syndrome.J Mol Cell Cardiol. 2015 Mar;80:186-95. doi: 10.1016/j.yjmcc.2015.01.002. Epub 2015 Jan 26. J Mol Cell Cardiol. 2015. PMID: 25633834
-
Cav1.2 channelopathies causing autism: new hallmarks on Timothy syndrome.Pflugers Arch. 2020 Jul;472(7):775-789. doi: 10.1007/s00424-020-02430-0. Epub 2020 Jul 3. Pflugers Arch. 2020. PMID: 32621084 Review.
-
CaV1.2 channelopathies: from arrhythmias to autism, bipolar disorder, and immunodeficiency.Pflugers Arch. 2010 Jul;460(2):353-9. doi: 10.1007/s00424-009-0753-0. Epub 2009 Nov 15. Pflugers Arch. 2010. PMID: 19916019 Review.
Cited by
-
CaV1.2 channelopathic mutations evoke diverse pathophysiological mechanisms.J Gen Physiol. 2022 Nov 7;154(11):e202213209. doi: 10.1085/jgp.202213209. Epub 2022 Sep 27. J Gen Physiol. 2022. PMID: 36167061 Free PMC article.
-
Targeted deep sequencing analyses of long QT syndrome in a Japanese population.PLoS One. 2022 Dec 8;17(12):e0277242. doi: 10.1371/journal.pone.0277242. eCollection 2022. PLoS One. 2022. PMID: 36480497 Free PMC article.
-
Multiple beta cell-independent mechanisms drive hypoglycemia in Timothy syndrome.Nat Commun. 2024 Oct 17;15(1):8980. doi: 10.1038/s41467-024-52885-3. Nat Commun. 2024. PMID: 39420001 Free PMC article.
-
The diagnosis and treatment of cardiac ion channelopathies: congenital long QT syndrome and Brugada syndrome.Curr Treat Options Cardiovasc Med. 2007 Oct;9(5):364-71. doi: 10.1007/s11936-007-0056-8. Curr Treat Options Cardiovasc Med. 2007. PMID: 17897565
-
Age and gender-dependent alternative splicing of P/Q-type calcium channel EF-hand.Neuroscience. 2007 Mar 30;145(3):1026-36. doi: 10.1016/j.neuroscience.2006.12.054. Epub 2007 Feb 8. Neuroscience. 2007. PMID: 17291689 Free PMC article.
References
-
- Reichenbach, H., Meister, E. M. & Theile, H. (1992) Kinderarztl Prax 60, 54-56. - PubMed
-
- Marks, M. L., Trippel, D. L. & Keating, M. T. (1995) Am. J. Cardiol. 76, 744-745. - PubMed
-
- Marks, M. L., Whisler, S. L., Clericuzio, C. & Keating, M. (1995) J. Am. Coll. Cardiol. 25, 59-64. - PubMed
-
- Splawski, I., Timothy, K. W., Sharpe, L. M., Decher, N., Kumar, P., Bloise, R., Napolitano, C., Schwartz, P. J., Joseph, R. M., Condouris, K., et al. (2004) Cell 119, 19-31. - PubMed
-
- Sanoudou, D. & Beggs, A. H. (2001) Trends Mol. Med. 7, 362-368. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
