

Syndromic patent ductus arteriosus: evidence for haploinsufficient TFAP2B mutations and identification of a linked sleep disorder
- PMID: 15684060
- PMCID: PMC549488
- DOI: 10.1073/pnas.0409852102
Syndromic patent ductus arteriosus: evidence for haploinsufficient TFAP2B mutations and identification of a linked sleep disorder
Abstract
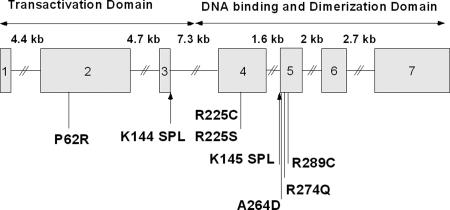
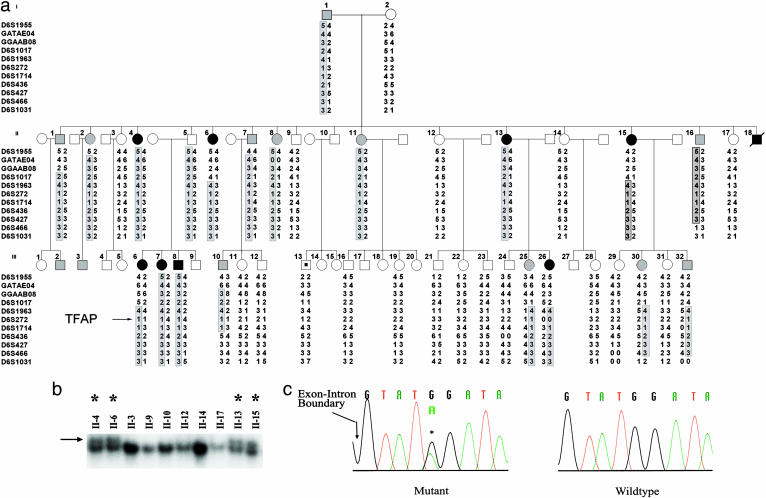
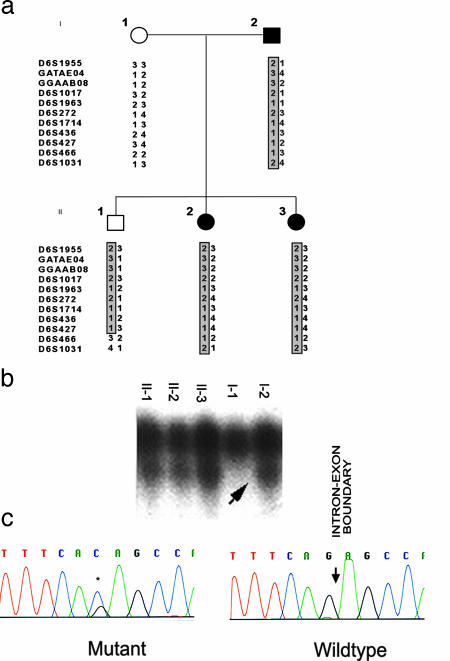
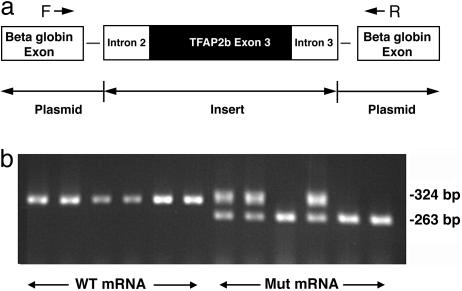
Patent ductus arteriosus (PDA) is a common congenital heart disease that results when the ductus arteriosus, a muscular artery, fails to remodel and close after birth. A syndromic form of this disorder, Char syndrome, is caused by mutation in TFAP2B, the gene encoding a neural crest-derived transcription factor. Established features of the syndrome are PDA, facial dysmorphology, and fifth-finger clinodactyly. Disease-causing mutations are missense and are proposed to be dominant negative. Because only a small number of families have been reported, there is limited information on the spectrum of mutations and resulting phenotypes. We report the characterization of two kindreds (K144 and K145) with Char syndrome containing 22 and 5 affected members, respectively. Genotyping revealed linkage to TFAP2B in both families. Sequencing of TFAP2B demonstrated mutations in both kindreds that were not found among control chromosomes. Both mutations altered highly conserved bases in introns required for normal splicing as demonstrated by biochemical studies in mammalian cells. The abnormal splicing results in mRNAs containing frameshift mutations that are expected to be degraded by nonsense-mediated mRNA decay, resulting in haploinsufficiency; even if produced, the protein in K144 would lack DNA binding and dimerization motifs and would likely result in haploinsufficiency. Examination of these two kindreds for phenotypes that segregate with TFAP2B mutations identified several phenotypes not previously linked to Char syndrome. These include parasomnia and dental and occipital-bone abnormalities. The striking sleep disorder in these kindreds implicates TFAP2B-dependent functions in the normal regulation of sleep.
Figures
Similar articles
-
Mutations in TFAP2B cause Char syndrome, a familial form of patent ductus arteriosus.Nat Genet. 2000 May;25(1):42-6. doi: 10.1038/75578. Nat Genet. 2000. PMID: 10802654
-
Familial nonsyndromic patent ductus arteriosus caused by mutations in TFAP2B.Pediatr Cardiol. 2011 Oct;32(7):958-65. doi: 10.1007/s00246-011-0024-7. Epub 2011 Jun 4. Pediatr Cardiol. 2011. PMID: 21643846
-
Characterization of transcription factor AP-2 β mutations involved in familial isolated patent ductus arteriosus suggests haploinsufficiency.J Surg Res. 2014 May 15;188(2):466-472. doi: 10.1016/j.jss.2014.01.015. Epub 2014 Jan 12. J Surg Res. 2014. PMID: 24507797 Free PMC article.
-
Molecular determinants of atrial and ventricular septal defects and patent ductus arteriosus.Am J Med Genet. 2000 Winter;97(4):304-9. doi: 10.1002/1096-8628(200024)97:4<304::aid-ajmg1281>3.0.co;2-#. Am J Med Genet. 2000. PMID: 11376442 Review.
-
[Research progress of genetic research on Char syndrome].Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2024 Jun 10;41(6):758-760. doi: 10.3760/cma.j.cn511374-20210607-00478. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2024. PMID: 38818565 Review. Chinese.
Cited by
-
Genetic basis of congenital cardiovascular malformations.Eur J Med Genet. 2014 Aug;57(8):402-13. doi: 10.1016/j.ejmg.2014.04.010. Epub 2014 Apr 30. Eur J Med Genet. 2014. PMID: 24793338 Free PMC article. Review.
-
Sleep Architecture in Mice Is Shaped by the Transcription Factor AP-2β.Genetics. 2020 Nov;216(3):753-764. doi: 10.1534/genetics.120.303435. Epub 2020 Sep 2. Genetics. 2020. PMID: 32878901 Free PMC article.
-
Crucial role of TFAP2B in the nervous system for regulating NREM sleep.Mol Brain. 2024 Feb 27;17(1):13. doi: 10.1186/s13041-024-01084-8. Mol Brain. 2024. PMID: 38413970 Free PMC article.
-
Frontal nasal prominence expression driven by Tcfap2a relies on a conserved binding site for STAT proteins.Dev Dyn. 2006 May;235(5):1358-70. doi: 10.1002/dvdy.20722. Dev Dyn. 2006. PMID: 16502414 Free PMC article.
-
Sleep-active neuron specification and sleep induction require FLP-11 neuropeptides to systemically induce sleep.Elife. 2016 Mar 7;5:e12499. doi: 10.7554/eLife.12499. Elife. 2016. PMID: 26949257 Free PMC article.
References
-
- Mitchell, S. C., Korones, S. B. & Berendes, H. W. (1971) Circulation 43, 323-332. - PubMed
-
- Hoffman, J. I. & Christianson, R. (1978) Am. J. Cardiol. 42, 641-647. - PubMed
-
- Ferencz, C., Rubin, J. D., McCarter, R. J., Brenner, J. I., Neill, C. A., Perry, L. W., Hepner, S. I. & Downing, J. W. (1985) Am. J. Epidemiol. 121, 31-36. - PubMed
-
- Gittenberger-de Groot, A. C. & Strengers, J. L. (1988) Int. J. Cardiol. 19, 153-166. - PubMed
-
- Gittenberger-de Groot, A. C., Strengers, J. L., Mentink, M., Poelmann, R. E. & Patterson, D. F. (1985) J. Am. Coll. Cardiol. 6, 394-404. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
