

Heterozygous TGFBR2 mutations in Marfan syndrome
- PMID: 15235604
- PMCID: PMC2230615
- DOI: 10.1038/ng1392
Heterozygous TGFBR2 mutations in Marfan syndrome
Abstract
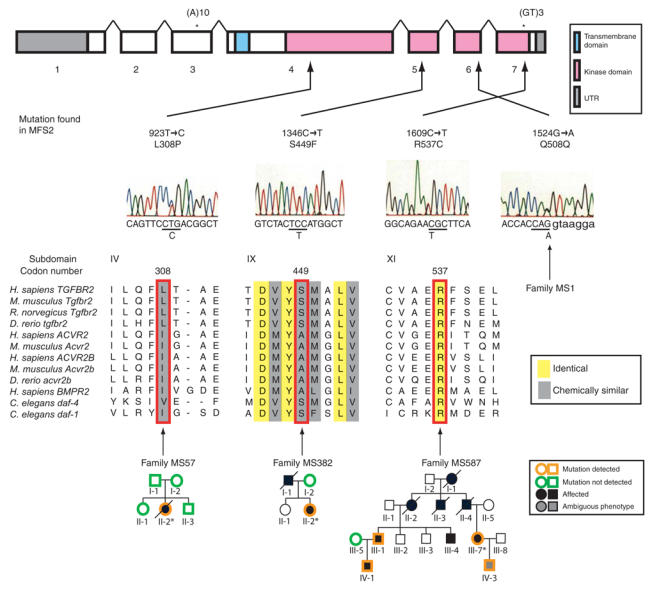
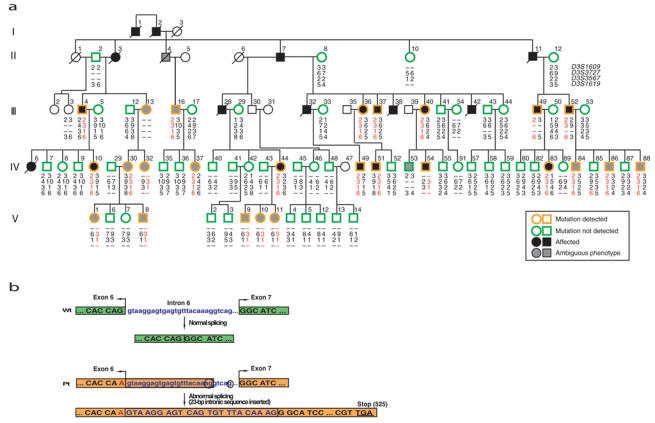
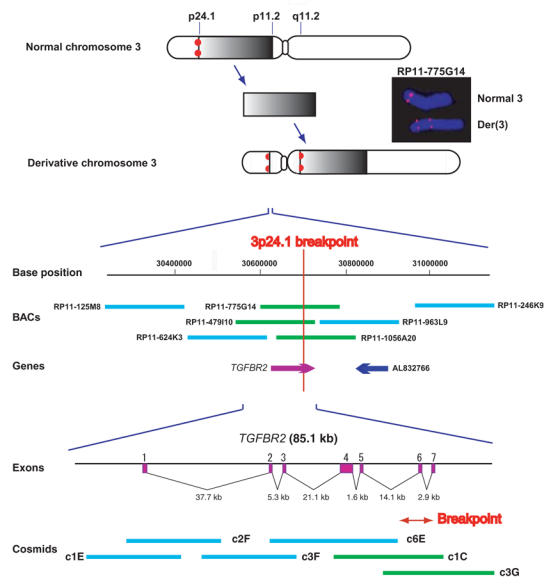
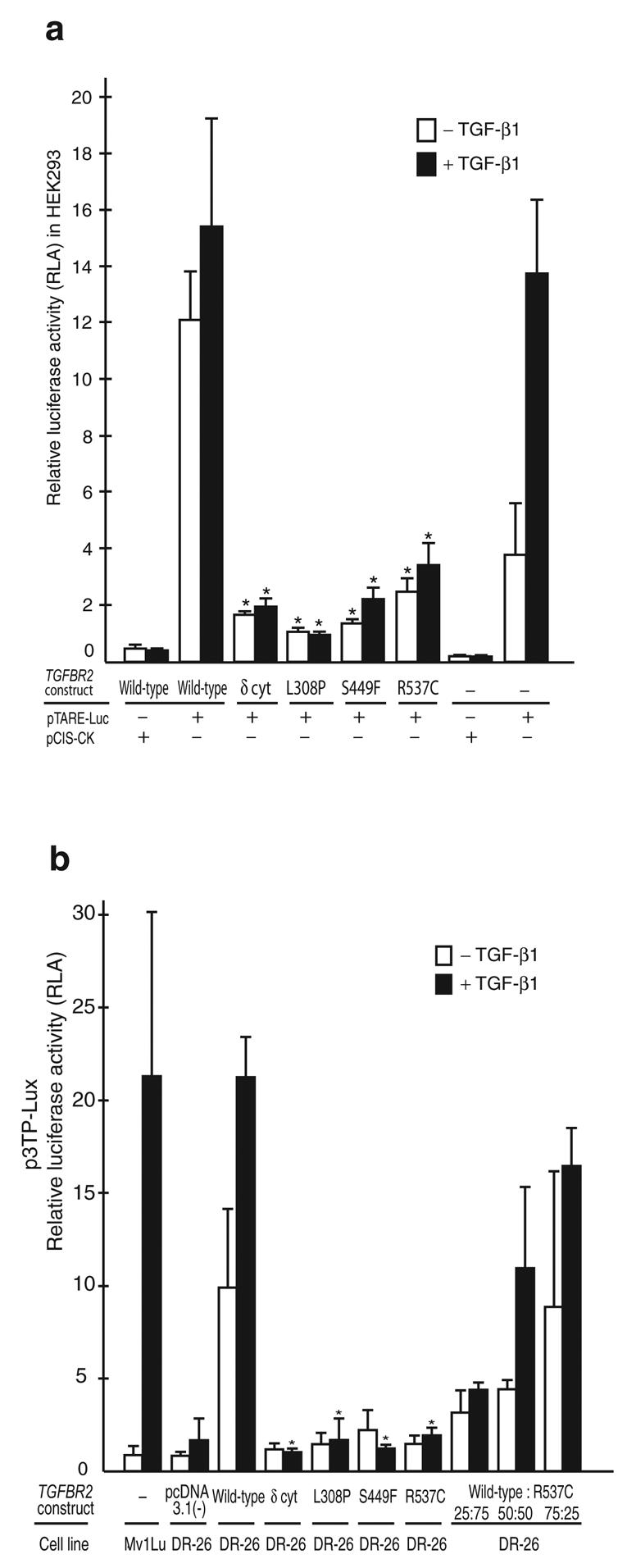
Marfan syndrome is an extracellular matrix disorder with cardinal manifestations in the eye, skeleton and cardiovascular systems associated with defects in the gene encoding fibrillin (FBN1) at 15q21.1 (ref. 1). A second type of the disorder (Marfan syndrome type 2; OMIM 154705) is associated with a second locus, MFS2, at 3p25-p24.2 in a large French family (family MS1). Identification of a 3p24.1 chromosomal breakpoint disrupting the gene encoding TGF-beta receptor 2 (TGFBR2) in a Japanese individual with Marfan syndrome led us to consider TGFBR2 as the gene underlying association with Marfan syndrome at the MSF2 locus. The mutation 1524G-->A in TGFBR2 (causing the synonymous amino acid substitution Q508Q) resulted in abnormal splicing and segregated with MFS2 in family MS1. We identified three other missense mutations in four unrelated probands, which led to loss of function of TGF-beta signaling activity on extracellular matrix formation. These results show that heterozygous mutations in TGFBR2, a putative tumor-suppressor gene implicated in several malignancies, are also associated with inherited connective-tissue disorders.
Figures
Comment in
-
TGF beta signaling in health and disease.Nat Genet. 2004 Aug;36(8):790-2. doi: 10.1038/ng0804-790. Nat Genet. 2004. PMID: 15284845 No abstract available.
-
Identification of a novel TGFBR2 gene mutation in a Korean patient with Loeys-Dietz aortic aneurysm syndrome; no mutation in TGFBR2 gene in 30 patients with classic Marfan's syndrome.Clin Genet. 2005 Dec;68(6):561-3. doi: 10.1111/j.1399-0004.2005.00535.x. Clin Genet. 2005. PMID: 16283890 No abstract available.
Similar articles
-
TGFBR1 and TGFBR2 mutations in patients with features of Marfan syndrome and Loeys-Dietz syndrome.Hum Mutat. 2006 Aug;27(8):770-7. doi: 10.1002/humu.20354. Hum Mutat. 2006. PMID: 16799921
-
Quantitative analysis of TGFBR2 mutations in Marfan-syndrome-related disorders suggests a correlation between phenotypic severity and Smad signaling activity.J Cell Sci. 2010 Dec 15;123(Pt 24):4340-50. doi: 10.1242/jcs.074773. Epub 2010 Nov 23. J Cell Sci. 2010. PMID: 21098638
-
Identification of 23 TGFBR2 and 6 TGFBR1 gene mutations and genotype-phenotype investigations in 457 patients with Marfan syndrome type I and II, Loeys-Dietz syndrome and related disorders.Hum Mutat. 2008 Nov;29(11):E284-95. doi: 10.1002/humu.20871. Hum Mutat. 2008. PMID: 18781618
-
Molecular genetics of Marfan syndrome.Curr Opin Cardiol. 2005 May;20(3):194-200. doi: 10.1097/01.hco.0000162398.21972.cd. Curr Opin Cardiol. 2005. PMID: 15861007 Review.
-
Marfan syndrome: from molecular pathogenesis to clinical treatment.Curr Opin Genet Dev. 2007 Jun;17(3):252-8. doi: 10.1016/j.gde.2007.04.006. Epub 2007 Apr 27. Curr Opin Genet Dev. 2007. PMID: 17467262 Review.
Cited by
-
Angiotensin-converting enzyme I/D polymorphism and the risk of thoracic aortic dissection in Chinese Han population.Mol Biol Rep. 2013 Feb;40(2):1249-54. doi: 10.1007/s11033-012-2167-x. Epub 2012 Oct 19. Mol Biol Rep. 2013. PMID: 23079706
-
Heterozygous modulation of TGF-β signaling does not influence Müller glia cell reactivity or proliferation following NMDA-induced damage.Histochem Cell Biol. 2015 Nov;144(5):443-55. doi: 10.1007/s00418-015-1354-y. Epub 2015 Jul 28. Histochem Cell Biol. 2015. PMID: 26215132
-
Molecular mechanisms of MMP9 overexpression and its role in emphysema pathogenesis of Smad3-deficient mice.Am J Physiol Lung Cell Mol Physiol. 2012 Jul;303(2):L89-96. doi: 10.1152/ajplung.00060.2012. Epub 2012 May 18. Am J Physiol Lung Cell Mol Physiol. 2012. PMID: 22610349 Free PMC article.
-
Role of mechanotransduction in vascular biology: focus on thoracic aortic aneurysms and dissections.Circ Res. 2015 Apr 10;116(8):1448-61. doi: 10.1161/CIRCRESAHA.114.304936. Circ Res. 2015. PMID: 25858068 Free PMC article. Review.
-
Management of an elderly patient with nonsyndromic TGFBR1-related aortopathy: A case report.Clin Case Rep. 2024 Aug 8;12(8):e9317. doi: 10.1002/ccr3.9317. eCollection 2024 Aug. Clin Case Rep. 2024. PMID: 39130808 Free PMC article.
References
-
- Hasham S, et al. Mapping a locus for familial thoracic aortic aneurysms and dissections (TAAD2) to 3p24–25. Circulation. 2003;107:3184–3190. - PubMed
-
- Grady W, et al. Mutational inactivation of transforming growth factor β receptor type II in microsatellite stable colon cancers. Cancer Res. 1999;59:320–324. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
