

Myotonic dystrophy: RNA pathogenesis comes into focus
- PMID: 15065017
- PMCID: PMC1181975
- DOI: 10.1086/383590
Myotonic dystrophy: RNA pathogenesis comes into focus
Abstract
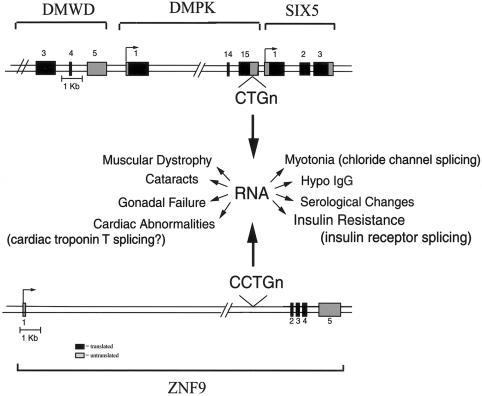
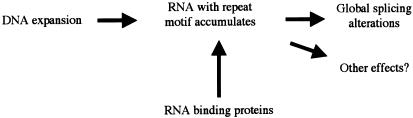
Myotonic dystrophy (DM)--the most common form of muscular dystrophy in adults, affecting 1/8000 individuals--is a dominantly inherited disorder with a peculiar and rare pattern of multisystemic clinical features affecting skeletal muscle, the heart, the eye, and the endocrine system. Two genetic loci have been associated with the DM phenotype: DM1, on chromosome 19, and DM2, on chromosome 3. In 1992, the mutation responsible for DM1 was identified as a CTG expansion located in the 3' untranslated region of the dystrophia myotonica-protein kinase gene (DMPK). How this untranslated CTG expansion causes myotonic dystrophy type 1(DM1) has been controversial. The recent discovery that myotonic dystrophy type 2 (DM2) is caused by an untranslated CCTG expansion, along with other discoveries on DM1 pathogenesis, indicate that the clinical features common to both diseases are caused by a gain-of-function RNA mechanism in which the CUG and CCUG repeats alter cellular function, including alternative splicing of various genes. We discuss the pathogenic mechanisms that have been proposed for the myotonic dystrophies, the clinical and molecular features of DM1 and DM2, and the characterization of murine and cell-culture models that have been generated to better understand these diseases.
Figures
Similar articles
-
Myotonic dystrophy: clinical and molecular parallels between myotonic dystrophy type 1 and type 2.Curr Neurol Neurosci Rep. 2002 Sep;2(5):465-70. doi: 10.1007/s11910-002-0074-6. Curr Neurol Neurosci Rep. 2002. PMID: 12169228 Review.
-
[Myotonic dystrophy].Nihon Rinsho. 2005 Mar;63(3):429-33. Nihon Rinsho. 2005. PMID: 15773341 Review. Japanese.
-
[Myotonic dystrophy - a new insight into a well-known disease].Neurol Neurochir Pol. 2010 May-Jun;44(3):264-76. doi: 10.1016/s0028-3843(14)60041-4. Neurol Neurochir Pol. 2010. PMID: 20625963 Review. Polish.
-
[Molecular pathways to myotonic dystrophy].Nihon Rinsho. 2005 Mar;63(3):515-21. Nihon Rinsho. 2005. PMID: 15773354 Review. Japanese.
-
Pathogenic mechanisms of myotonic dystrophy.Biochem Soc Trans. 2009 Dec;37(Pt 6):1281-6. doi: 10.1042/BST0371281. Biochem Soc Trans. 2009. PMID: 19909263 Free PMC article. Review.
Cited by
-
Structural characterization of naturally occurring RNA single mismatches.Nucleic Acids Res. 2011 Feb;39(3):1081-94. doi: 10.1093/nar/gkq793. Epub 2010 Sep 28. Nucleic Acids Res. 2011. PMID: 20876693 Free PMC article.
-
On the Applicability of Elastic Network Models for the Study of RNA CUG Trinucleotide Repeat Overexpansion.PLoS One. 2016 Mar 24;11(3):e0152049. doi: 10.1371/journal.pone.0152049. eCollection 2016. PLoS One. 2016. PMID: 27010216 Free PMC article.
-
Beyond the binding site: in vivo identification of tbx2, smarca5 and wnt5b as molecular targets of CNBP during embryonic development.PLoS One. 2013 May 7;8(5):e63234. doi: 10.1371/journal.pone.0063234. Print 2013. PLoS One. 2013. PMID: 23667590 Free PMC article.
-
Premature senescence in primary muscle cultures of myotonic dystrophy type 2 is not associated with p16 induction.Eur J Histochem. 2014 Oct 22;58(4):2444. doi: 10.4081/ejh.2014.2444. Eur J Histochem. 2014. PMID: 25578974 Free PMC article.
-
Epiretinal membrane: a treatable cause of visual disability in myotonic dystrophy type 1.J Neurol. 2014 Jan;261(1):37-44. doi: 10.1007/s00415-013-7141-6. Epub 2013 Oct 17. J Neurol. 2014. PMID: 24132671
References
Electronic-Database Information
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
-
- Bachinski LL, Udd B, Meola G, Sansone V, Bassez G, Eymard B, Thornton CA, Moxley RT, Harper PS, Rogers MT, Jurkat-Rott K, Lehmann-Horn F, Wieser T, Gamez J, Navarro C, Bottani A, Kohler A, Shriver MD, Sallinen R, Wessman M, Zhang S, Wright FA, Krahe R (2003) Confirmation of the type 2 myotonic dystrophy (CCTG)n expansion mutation in patients with proximal myotonic myopathy/proximal myotonic dystrophy of different European origins: a single shared haplotype indicates an ancestral founder effect. Am J Hum Genet 73:835–848 - PMC - PubMed
-
- Boucher CA, King SK, Carey N, Krahe R, Winchester CL, Rahman S, Creavin T, Meghji P, Bailey MES, Chartier FL, Brown SD, Siciliano MJ, Johnson KJ (1995) A novel homeodomain-encoding gene is associated with a large CpG island interrupted by the myotonic dystrophy unstable (CTG)n repeat. Hum Mol Genet 4:1919–1925 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
