

Mutations in a gene encoding a novel SH3/TPR domain protein cause autosomal recessive Charcot-Marie-Tooth type 4C neuropathy
- PMID: 14574644
- PMCID: PMC1180490
- DOI: 10.1086/379525
Mutations in a gene encoding a novel SH3/TPR domain protein cause autosomal recessive Charcot-Marie-Tooth type 4C neuropathy
Abstract
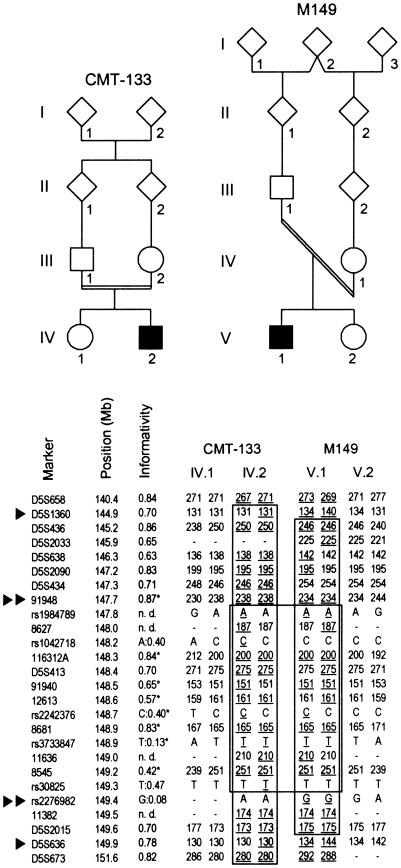
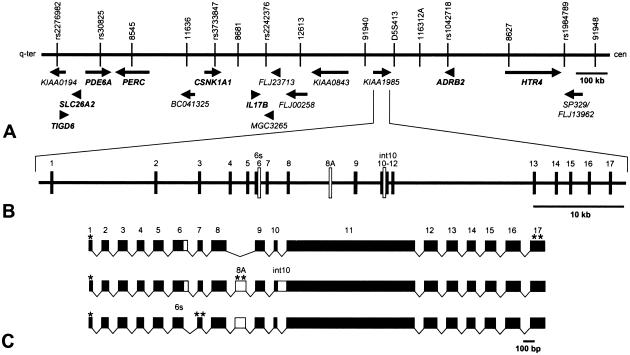
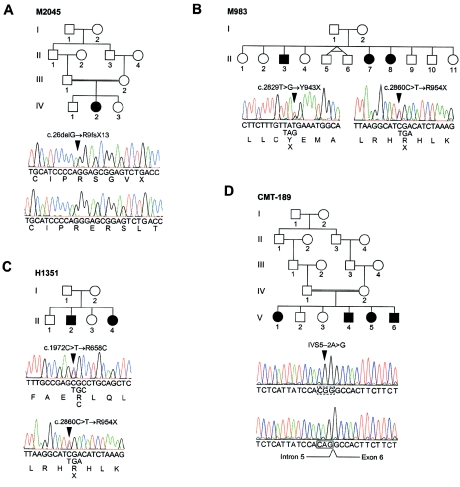
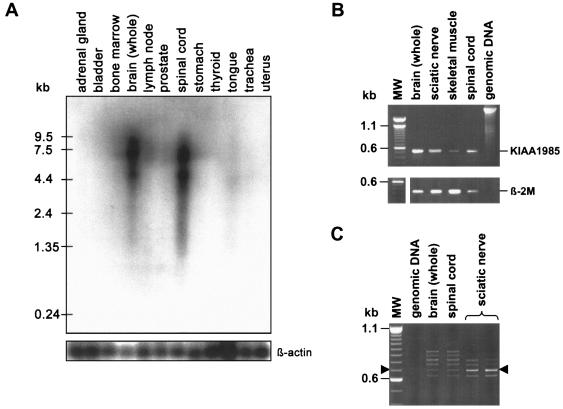
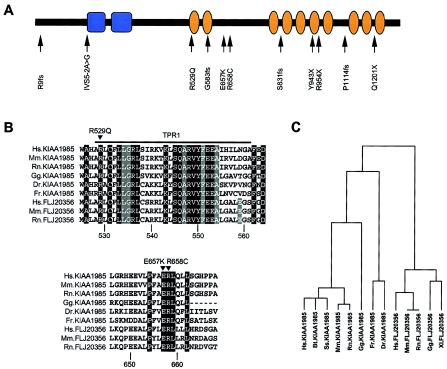
Charcot-Marie-Tooth disease type 4C (CMT4C) is a childhood-onset demyelinating form of hereditary motor and sensory neuropathy associated with an early-onset scoliosis and a distinct Schwann cell pathology. CMT4C is inherited as an autosomal recessive trait and has been mapped to a 13-cM linkage interval on chromosome 5q23-q33. By homozygosity mapping and allele-sharing analysis, we refined the CMT4C locus to a suggestive critical region of 1.7 Mb. We subsequently identified mutations in an uncharacterized transcript, KIAA1985, in 12 families with autosomal recessive neuropathy. We observed eight distinct protein-truncating mutations and three nonconservative missense mutations affecting amino acids conserved through evolution. In all families, we identified a mutation on each disease allele, either in the homozygous or in the compound heterozygous state. The CMT4C gene is strongly expressed in neural tissues, including peripheral nerve tissue. The translated protein defines a new protein family of unknown function with putative orthologues in vertebrates. Comparative sequence alignments indicate that members of this protein family contain multiple SH3 and TPR domains that are likely involved in the formation of protein complexes.
Figures



Similar articles
-
Study on the gene and phenotypic characterisation of autosomal recessive demyelinating motor and sensory neuropathy (Charcot-Marie-Tooth disease) with a gene locus on chromosome 5q23-q33.J Neurol Neurosurg Psychiatry. 1999 May;66(5):569-74. doi: 10.1136/jnnp.66.5.569. J Neurol Neurosurg Psychiatry. 1999. PMID: 10209165 Free PMC article.
-
The phenotype of Charcot-Marie-Tooth disease type 4C due to SH3TC2 mutations and possible predisposition to an inflammatory neuropathy.Neuromuscul Disord. 2009 Apr;19(4):264-9. doi: 10.1016/j.nmd.2009.01.006. Epub 2009 Mar 9. Neuromuscul Disord. 2009. PMID: 19272779
-
Spine deformities in Charcot-Marie-Tooth 4C caused by SH3TC2 gene mutations.Neurology. 2006 Aug 22;67(4):602-6. doi: 10.1212/01.wnl.0000230225.19797.93. Neurology. 2006. PMID: 16924012
-
[The genetics of type 1 Charcot-Marie-Tooth disease, the hereditary focal neuropathies and the hereditary distal motor neuropathies].Rev Neurol. 2000 Jan 1-15;30(1):71-9. Rev Neurol. 2000. PMID: 10743001 Review. Spanish.
-
[Molecular genetics of inherited neuropathies].Rinsho Shinkeigaku. 2006 Jan;46(1):1-18. Rinsho Shinkeigaku. 2006. PMID: 16541790 Review. Japanese.
Cited by
-
Clinical and electrophysiological aspects of Charcot-Marie-Tooth disease.Neuromolecular Med. 2006;8(1-2):3-22. doi: 10.1385/nmm:8:1-2:3. Neuromolecular Med. 2006. PMID: 16775364 Review.
-
Haplotype-specific modulation of a SOX10/CREB response element at the Charcot-Marie-Tooth disease type 4C locus SH3TC2.Hum Mol Genet. 2014 Oct 1;23(19):5171-87. doi: 10.1093/hmg/ddu240. Epub 2014 May 15. Hum Mol Genet. 2014. PMID: 24833716 Free PMC article.
-
Gene Therapy Options as New Treatment for Inherited Peripheral Neuropathy.Exp Neurobiol. 2020 Jun 30;29(3):177-188. doi: 10.5607/en20004. Exp Neurobiol. 2020. PMID: 32624504 Free PMC article. Review.
-
Exclusive expression of the Rab11 effector SH3TC2 in Schwann cells links integrin-α6 and myelin maintenance to Charcot-Marie-Tooth disease type 4C.Biochim Biophys Acta. 2016 Jul;1862(7):1279-90. doi: 10.1016/j.bbadis.2016.04.003. Epub 2016 Apr 9. Biochim Biophys Acta. 2016. PMID: 27068304 Free PMC article.
-
Neuropathology of Charcot-Marie-Tooth and related disorders.Neuromolecular Med. 2006;8(1-2):23-42. doi: 10.1385/nmm:8:1-2:23. Neuromolecular Med. 2006. PMID: 16775365 Review.
References
Electronic-Database Information
-
- Applied Biosystems SNP genotyping repository, http://myscience.appliedbiosystems.com/navigation/mysciLoginTC.jsp/ (for the publicly accessible section)
-
- Ensembl Genome Browser, http://www.ensembl.org/Multi/blastview?species=danio_rerio/ and http://www.ensembl.org/Multi/blastview?species=Fugu_rubripes/ (for BLAST searches on D. rerio and F. rubripes genomes, respectively)
-
- European Bioinformatics Institute Web site, http://www.ebi.ac.uk/index.html (for ClustalW server and Jalview)
-
- FGENESH, http://www.softberry.com/berry.phtml?topic=fgenesh&group=programs&subgro... (for ab initio gene prediction)
References
-
- Azzedine H, Bolino A, Taïeb T, Birouk N, Di Duca M, Bouhouche A, Benamou S, Mrabet A, Hammadouche T, Chkili T, Gouider R, Ravazzolo R, Brice A, Laporte J, LeGuern E (2003) Mutations in MTMR13, a new pseudophosphatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive demyelinating form of Charcot-Marie-Tooth disease associated with early-onset glaucoma. Am J Hum Genet 72:1141–1153 - PMC - PubMed
-
- Baxter RV, Ben Othmane K, Rochelle JM, Stajich JE, Hulette C, Dew-Knight S, Hentati F, Ben Hamida M, Bel S, Stenger JE, Gilbert JR, Pericak-Vance MA, Vance JM (2002) Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat Genet 30:21–22 - PubMed
-
- Bingle CD, Craig RW, Swales BM, Singleton V, Zhou P, Whyte MK (2000) Exon skipping in Mcl-1 results in a bcl-2 homology domain 3 only gene product that promotes cell death. J Biol Chem 275:22136–22146 - PubMed
-
- Blatch GL, Lässle M (1999) The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays 21:932–939 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
