

Barbiturates induce mitochondrial depolarization and potentiate excitotoxic neuronal death
- PMID: 12417645
- PMCID: PMC6758030
- DOI: 10.1523/JNEUROSCI.22-21-09203.2002
Barbiturates induce mitochondrial depolarization and potentiate excitotoxic neuronal death
Abstract
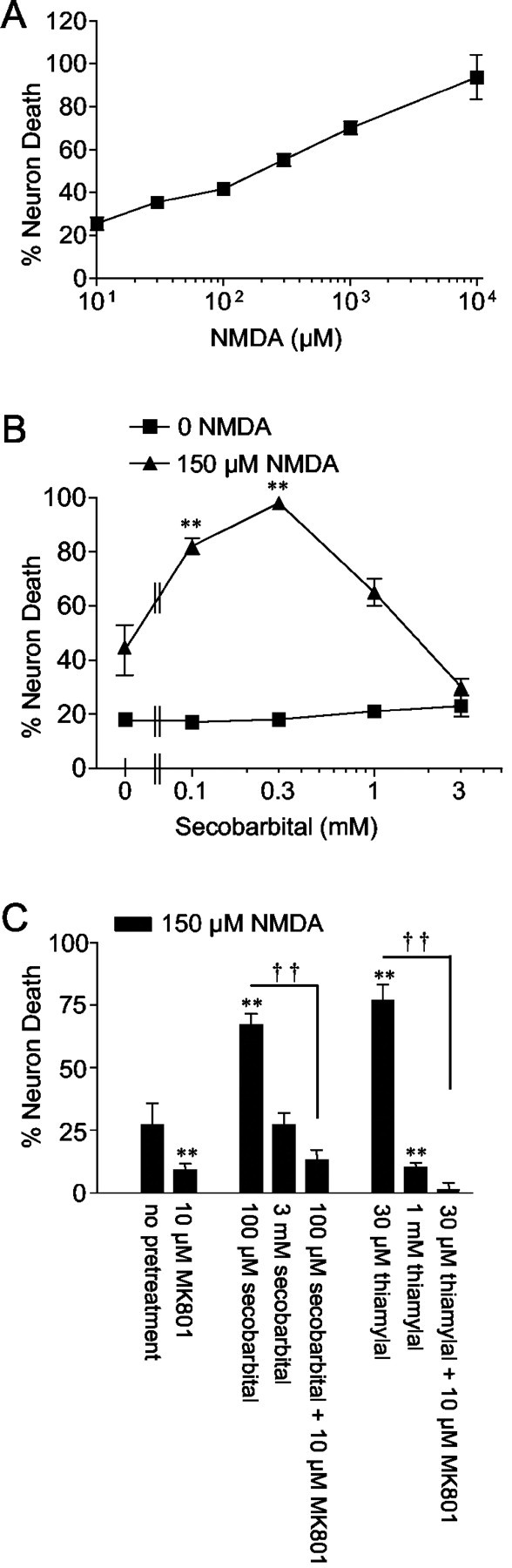
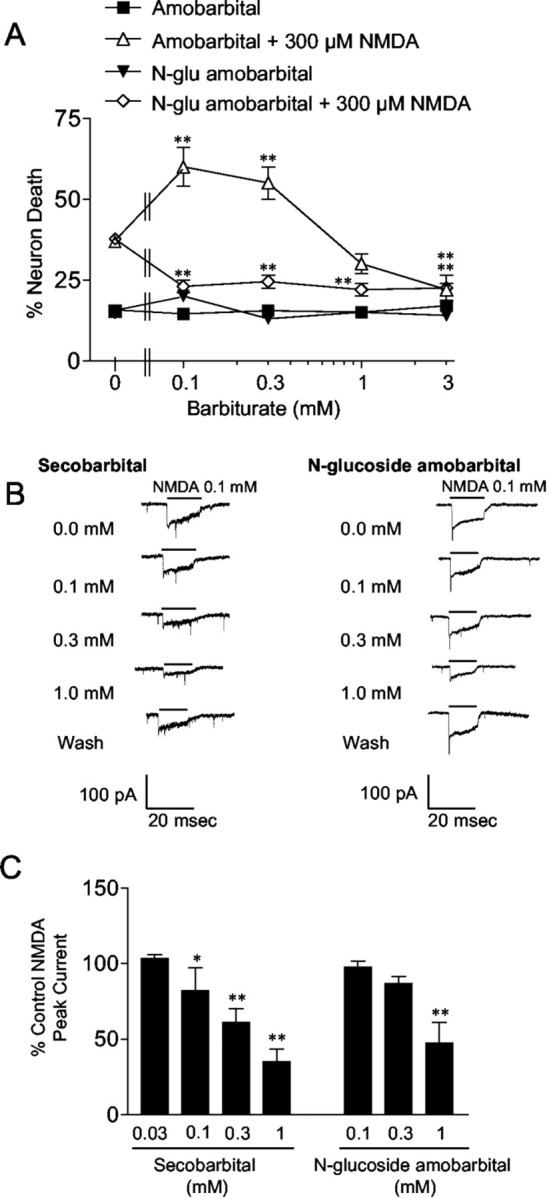
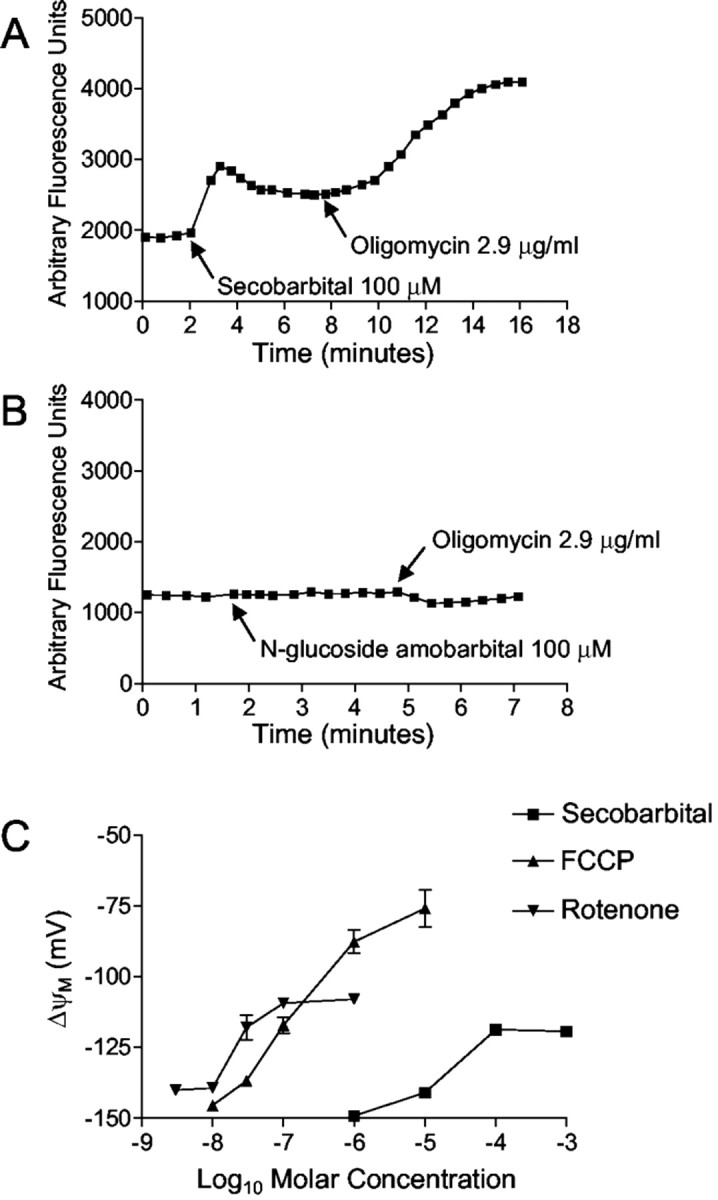
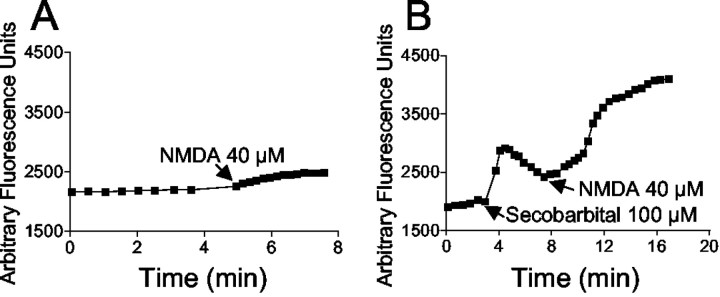
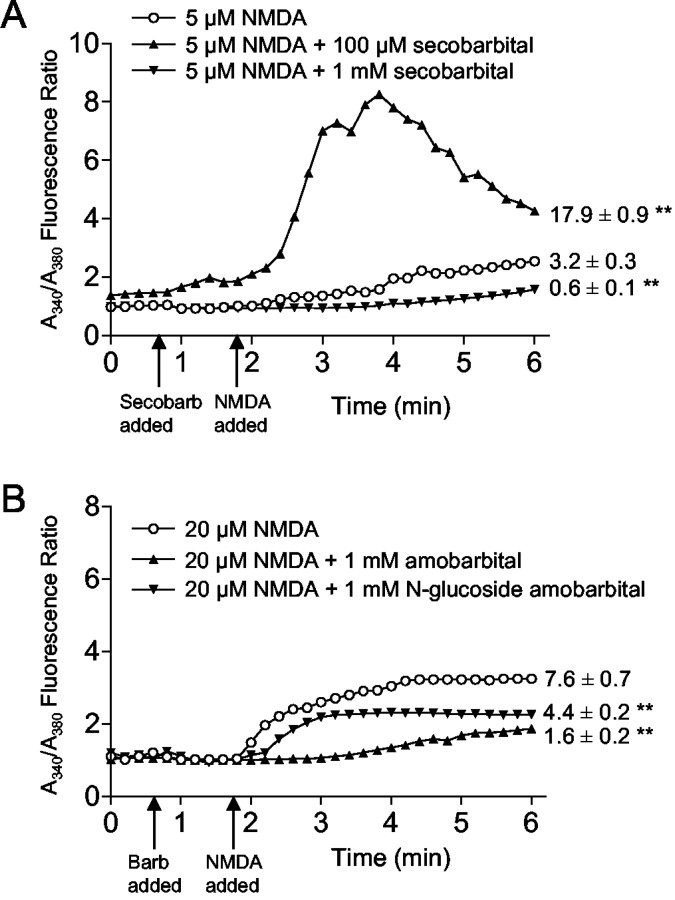
Barbiturates are widely used as anesthetics, anticonvulsants, and neuroprotective agents. However, barbiturates may also inhibit mitochondrial respiration, and mitochondrial inhibitors are known to potentiate NMDA receptor-mediated neurotoxicity. Here we used rat cortical cultures to examine the effect of barbiturates on neuronal mitochondria and responses to NMDA receptor stimulation. The barbiturates tested, secobarbital, amobarbital, and thiamylal, each potentiated NMDA-induced neuron death at barbiturate concentrations relevant to clinical and experimental use (100-300 microm). By using rhodamine-123 under quenching conditions, barbiturates in this concentration range were shown to depolarize neuronal mitochondria and greatly amplify NMDA-induced mitochondrial depolarization. Barbiturate-induced mitochondrial depolarization was increased by the ATP synthase inhibitor oligomycin, indicating that barbiturates act by inhibiting electron transport sufficiently to cause ATP synthase reversal. Barbiturates similarly amplified the effects of NMDA on cytoplasmic free calcium concentrations. The cell-impermeant barbiturate N-glucoside amobarbital did not influence mitochondrial potential or potentiate NMDA neurotoxicity or calcium responses. However, all of the barbiturates attenuated NMDA-induced calcium elevations and cell death when present at millimolar concentrations. Whole-cell patch-clamp studies showed that these effects may be attributable to actions at the cell membrane, resulting in a block of NMDA-induced current flux at millimolar barbiturate concentrations. Together, these findings reconcile previous reports of opposing effects on barbiturates on NMDA neurotoxicity and show that barbiturate effects on neuronal mitochondria can be functionally significant. Effects of barbiturates on neuronal mitochondria should be considered in experimental and clinical application of these drugs.
Figures
Similar articles
-
Acute, chronic and differential effects of several anesthetic barbiturates on glutamate receptor activation in neuronal culture.Brain Res. 1993 May 21;611(2):181-6. doi: 10.1016/0006-8993(93)90501-d. Brain Res. 1993. PMID: 8334512
-
N-methyl-D-aspartate receptor-mediated mitochondrial Ca(2+) overload in acute excitotoxic motor neuron death: a mechanism distinct from chronic neurotoxicity after Ca(2+) influx.J Neurosci Res. 2001 Mar 1;63(5):377-87. doi: 10.1002/1097-4547(20010301)63:5<377::AID-JNR1032>3.0.CO;2-#. J Neurosci Res. 2001. PMID: 11223912
-
Effects of volatile anesthetics on N-methyl-D-aspartate excitotoxicity in primary rat neuronal-glial cultures.Anesthesiology. 2001 Sep;95(3):756-65. doi: 10.1097/00000542-200109000-00031. Anesthesiology. 2001. PMID: 11575551
-
Toward the development of strategies to prevent ischemic neuronal injury. In vitro studies.Ann N Y Acad Sci. 1997 Oct 15;825:209-19. doi: 10.1111/j.1749-6632.1997.tb48431.x. Ann N Y Acad Sci. 1997. PMID: 9369988 Review.
-
Mitochondria, glutamate neurotoxicity and the death cascade.Biochim Biophys Acta. 1998 Aug 10;1366(1-2):113-26. doi: 10.1016/s0005-2728(98)00124-8. Biochim Biophys Acta. 1998. PMID: 9714770 Review.
Cited by
-
Facial Dysmorphism, Hirsutism, and Failure to Thrive as Manifestation of Leigh Syndrome in a Child with SURF1 Mutation.J Pediatr Neurosci. 2020 Apr-Jun;15(2):108-110. doi: 10.4103/jpn.JPN_137_18. Epub 2020 Jun 27. J Pediatr Neurosci. 2020. PMID: 33042241 Free PMC article.
-
Inhibition of CorA-Dependent Magnesium Homeostasis Is Cidal in Mycobacterium tuberculosis.Antimicrob Agents Chemother. 2019 Sep 23;63(10):e01006-19. doi: 10.1128/AAC.01006-19. Print 2019 Oct. Antimicrob Agents Chemother. 2019. PMID: 31383669 Free PMC article.
-
Elementary Ca2+ release events in mammalian skeletal muscle: effects of the anaesthetic drug thiopental.J Muscle Res Cell Motil. 2006;27(5-7):315-26. doi: 10.1007/s10974-006-9092-3. Epub 2006 Aug 4. J Muscle Res Cell Motil. 2006. PMID: 16897573
-
The Effect of Ginsenoside RB1, Diazoxide, and 5-Hydroxydecanoate on Hypoxia-Reoxygenation Injury of H9C2 Cardiomyocytes.Evid Based Complement Alternat Med. 2019 Dec 13;2019:6046405. doi: 10.1155/2019/6046405. eCollection 2019. Evid Based Complement Alternat Med. 2019. PMID: 31915449 Free PMC article.
-
Mitochondrial inner membrane electrophysiology assessed by rhodamine-123 transport and fluorescence.Ann Biomed Eng. 2007 Jul;35(7):1276-85. doi: 10.1007/s10439-007-9265-2. Epub 2007 Mar 20. Ann Biomed Eng. 2007. PMID: 17372838 Free PMC article.
References
-
- Airey IL, Smith PA, Stoddart JC. Plasma and cerebrospinal fluid barbiturate levels during prolonged continuous thiopentone infusion. Anaesthesia. 1982;37:328–331. - PubMed
-
- Beal MF. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann Neurol. 1992;31:119–130. - PubMed
-
- Bhayana V, Alto LE, Dhalla NS. Effects of pentobarbital and pentothal on rat heart contractile force and oxidative phosphorylation activities. Gen Pharmacol. 1980;11:375–377. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources