

The molecular basis of X-linked spondyloepiphyseal dysplasia tarda
- PMID: 11349230
- PMCID: PMC1226125
- DOI: 10.1086/320592
The molecular basis of X-linked spondyloepiphyseal dysplasia tarda
Abstract
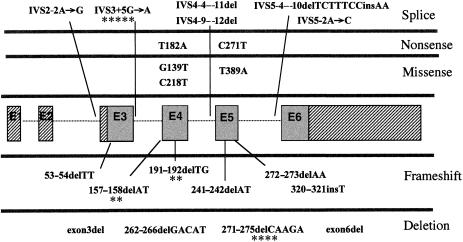
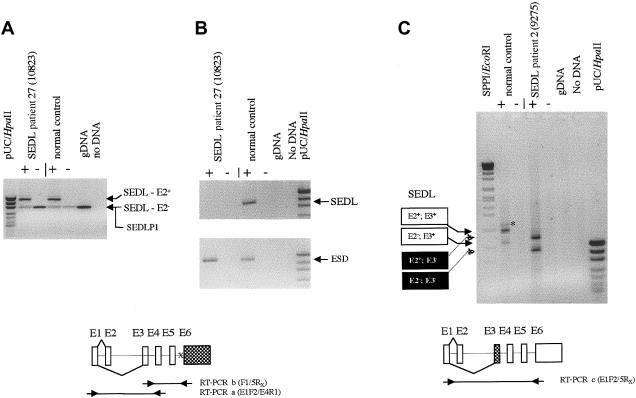
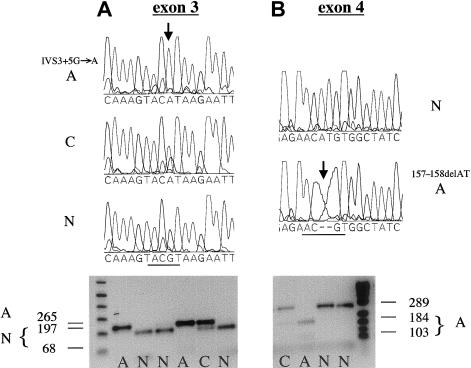
The X-linked form of spondyloepiphyseal dysplasia tarda (SEDL), a radiologically distinct skeletal dysplasia affecting the vertebrae and epiphyses, is caused by mutations in the SEDL gene. To characterize the molecular basis for SEDL, we have identified the spectrum of SEDL mutations in 30 of 36 unrelated cases of X-linked SEDL ascertained from different ethnic populations. Twenty-one different disease-associated mutations now have been identified throughout the SEDL gene. These include nonsense mutations in exons 4 and 5, missense mutations in exons 4 and 6, small (2-7 bp) and large (>1 kb) deletions, insertions, and putative splicing errors, with one splicing error due to a complex deletion/insertion mutation. Eight different frameshift mutations lead to a premature termination of translation and account for >43% (13/30) of SEDL cases, with half of these (7/13) being due to dinucleotide deletions. Altogether, deletions account for 57% (17/30) of all known SEDL mutations. Four recurrent mutations (IVS3+5G-->A, 157-158delAT, 191-192delTG, and 271-275delCAAGA) account for 43% (13/30) of confirmed SEDL cases. The results of haplotype analyses and the diverse ethnic origins of patients support recurrent mutations. Two patients with large deletions of SEDL exons were found, one with childhood onset of painful complications, the other relatively free of additional symptoms. However, we could not establish a clear genotype/phenotype correlation and therefore conclude that the complete unaltered SEDL-gene product is essential for normal bone growth. Molecular diagnosis can now be offered for presymptomatic testing of this disorder. Appropriate lifestyle decisions and, eventually, perhaps, specific SEDL therapies may ameliorate the prognosis of premature osteoarthritis and the need for hip arthroplasty.
Figures
Similar articles
-
A recurrent RNA-splicing mutation in the SEDL gene causes X-linked spondyloepiphyseal dysplasia tarda.Am J Hum Genet. 2001 Jun;68(6):1398-407. doi: 10.1086/320594. Epub 2001 Apr 26. Am J Hum Genet. 2001. PMID: 11326333 Free PMC article.
-
[Gene diagnosis of X-linked spondyloepiphyseal dysplasia tarda by linkage analysis and DNA sequencing].Zhonghua Er Ke Za Zhi. 2003 Apr;41(4):256-9. Zhonghua Er Ke Za Zhi. 2003. PMID: 14754526 Chinese.
-
Preonset studies of spondyloepiphyseal dysplasia tarda caused by a novel 2-base pair deletion in SEDL encoding sedlin.J Bone Miner Res. 2001 Dec;16(12):2245-50. doi: 10.1359/jbmr.2001.16.12.2245. J Bone Miner Res. 2001. PMID: 11760838
-
Identification of three novel SEDL mutations, including mutation in the rare, non-canonical splice site of exon 4.Clin Genet. 2003 Sep;64(3):235-42. doi: 10.1034/j.1399-0004.2003.00132.x. Clin Genet. 2003. PMID: 12919139 Review.
-
Novel and recurrent EBP mutations in X-linked dominant chondrodysplasia punctata.Am J Med Genet. 2000 Oct 2;94(4):300-5. doi: 10.1002/1096-8628(20001002)94:4<300::aid-ajmg7>3.0.co;2-3. Am J Med Genet. 2000. PMID: 11038443 Review.
Cited by
-
X-linked spondyloepiphyseal dysplasia tarda: Identification of a TRAPPC2 mutation in a Korean pedigree.Ann Lab Med. 2012 May;32(3):234-7. doi: 10.3343/alm.2012.32.3.234. Epub 2012 Apr 18. Ann Lab Med. 2012. PMID: 22563562 Free PMC article.
-
A recurrent RNA-splicing mutation in the SEDL gene causes X-linked spondyloepiphyseal dysplasia tarda.Am J Hum Genet. 2001 Jun;68(6):1398-407. doi: 10.1086/320594. Epub 2001 Apr 26. Am J Hum Genet. 2001. PMID: 11326333 Free PMC article.
-
C4orf41 and TTC-15 are mammalian TRAPP components with a role at an early stage in ER-to-Golgi trafficking.Mol Biol Cell. 2011 Jun 15;22(12):2083-93. doi: 10.1091/mbc.E10-11-0873. Epub 2011 Apr 27. Mol Biol Cell. 2011. PMID: 21525244 Free PMC article.
-
Novel loss-of-function variants of TRAPPC2 manifesting X-linked spondyloepiphyseal dysplasia tarda: report of two cases.BMC Med Genet. 2019 May 3;20(1):70. doi: 10.1186/s12881-019-0802-2. BMC Med Genet. 2019. PMID: 31053099 Free PMC article.
-
A novel RNA-splicing mutation in TRAPPC2 gene causing x-linked spondyloepiphyseal dysplasia tarda in a large Chinese family.J Genet. 2009 Apr;88(1):87-91. doi: 10.1007/s12041-009-0012-3. J Genet. 2009. PMID: 19417549 No abstract available.
References
Electronic-Database Information
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/index.html (for SEDL mRNA [accession number NM_014563] and SEDL exons 1 [accession number AF157060], 2 [accession number AF157061], 3 [accession number AF157062], 4 [accession number AF157065], 5 [accession number AF157064], and 6 [accession number AF157065])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for SEDT [MIM 271600, MIM 184100], SEDL[MIM 300202], and SEDL [SEDT, X-linked] [MIM 313400])
References
-
- Beighton P (1997) Hereditary noninflammatory arthropathies. In: Rimoin DL, Connor JM, Pyeritz RE (eds) Emery and Rimoin’s principles and practice of medical genetics, 3d ed. Vol II. Churchill Livingstone, New York, pp 2773–2777
-
- Bieganski T, Dawydzik B, Kozlowski K (1999) Spondylo-epimetaphyseal dysplasia: a new X-linked variant with mental retardation. Eur J Pediatr 158:809–814 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
