

Genetic basis of sitosterolemia
- PMID: 11264985
- PMCID: PMC1350992
- DOI: 10.1097/00041433-200104000-00007
Genetic basis of sitosterolemia
Abstract
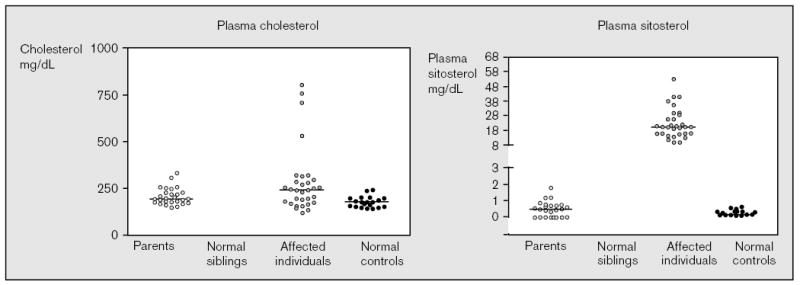
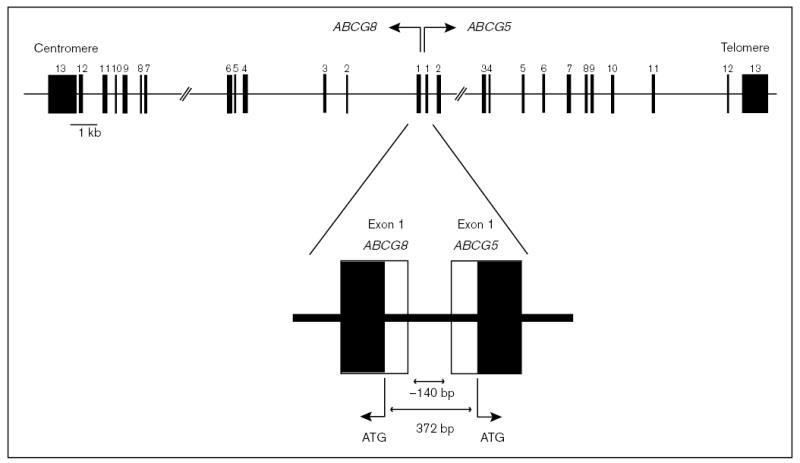
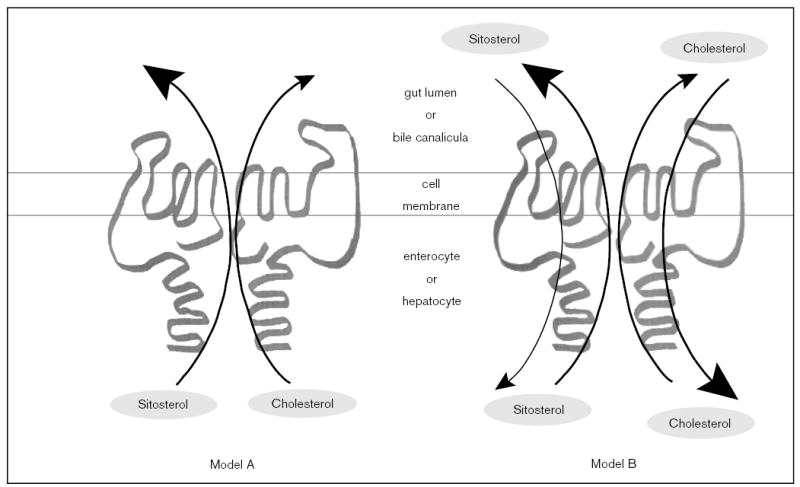
The molecular mechanisms regulating the amount of dietary cholesterol retained by the body, as well as the body's ability to exclude other dietary sterols selectively, are poorly understood. An average Western diet will contain approximately 250-500 mg of dietary cholesterol and approximately 200-400 mg of non-cholesterol sterols, of which plant sterols are the major constituents. Approximately 50-60% of dietary cholesterol is absorbed and retained by the normal human body, but less than 1% of the non-cholesterol sterols are retained. There thus exists a subtle mechanism that allows the body to distinguish between cholesterol and non-cholesterol sterols. In sitosterolemia, a rare autosomal recessive disorder, affected individuals hyperabsorb and retain not only cholesterol but also all other sterols, including plant and shellfish sterols from the intestine. Consequently, patients with this disease have very high levels of plant sterols in the plasma, and develop tendon and tuberous xanthomas, accelerated atherosclerosis, and premature coronary artery disease. The STSL locus has been mapped to human chromosome 2p21. Mutations in two tandem ABC genes, ABCG5 and ABCG8, encoding sterolin-1 and -2, respectively, are now known to be mutant in sitosterolemia. The identification of these genes should now lead to a better understanding of the molecular mechanism(s) governing the highly selective absorption and retention of cholesterol by the body. Indeed, it is the very existence of this disease that has given credence to the hypothesis that there is a molecular pathway that regulates dietary cholesterol absorption and sterol excretion by the body.
Figures
Similar articles
-
Identification of a gene, ABCG5, important in the regulation of dietary cholesterol absorption.Nat Genet. 2001 Jan;27(1):79-83. doi: 10.1038/83799. Nat Genet. 2001. PMID: 11138003 Free PMC article.
-
Two genes that map to the STSL locus cause sitosterolemia: genomic structure and spectrum of mutations involving sterolin-1 and sterolin-2, encoded by ABCG5 and ABCG8, respectively.Am J Hum Genet. 2001 Aug;69(2):278-90. doi: 10.1086/321294. Epub 2001 Jul 9. Am J Hum Genet. 2001. PMID: 11452359 Free PMC article.
-
Mapping a gene involved in regulating dietary cholesterol absorption. The sitosterolemia locus is found at chromosome 2p21.J Clin Invest. 1998 Sep 1;102(5):1041-4. doi: 10.1172/JCI3963. J Clin Invest. 1998. PMID: 9727073 Free PMC article.
-
Sitosterolaemia: pathophysiology, clinical presentation and laboratory diagnosis.J Clin Pathol. 2008 May;61(5):588-94. doi: 10.1136/jcp.2007.049775. J Clin Pathol. 2008. PMID: 18441155 Review.
-
Sitosterolemia.Cardiovasc Drug Rev. 2002 Winter;20(4):255-70. doi: 10.1111/j.1527-3466.2002.tb00096.x. Cardiovasc Drug Rev. 2002. PMID: 12481199 Review.
Cited by
-
Do plant sterol concentrations correlate with coronary artery disease in type 1 diabetes? A report from the Pittsburgh Epidemiology of Diabetes Complications Study.J Diabetes. 2009 Jun;1(2):112-7. doi: 10.1111/j.1753-0407.2009.00012.x. J Diabetes. 2009. PMID: 20827426 Free PMC article.
-
A Multiplex Phytosterol Assay Utilizing Gas Chromatography-Mass Spectrometry for Diagnosis of Inherited Lipid Storage Disorders.Ann Lab Med. 2019 Jul;39(4):411-413. doi: 10.3343/alm.2019.39.4.411. Ann Lab Med. 2019. PMID: 30809990 Free PMC article. No abstract available.
-
High prevalence of increased sitosterol levels in hypercholesterolemic children suggest underestimation of sitosterolemia incidence.PLoS One. 2020 Aug 26;15(8):e0238079. doi: 10.1371/journal.pone.0238079. eCollection 2020. PLoS One. 2020. PMID: 32845916 Free PMC article.
-
Emerging LDL therapies: Using human genetics to discover new therapeutic targets for plasma lipids.J Clin Lipidol. 2013 May-Jun;7(3 Suppl):S1-5. doi: 10.1016/j.jacl.2013.03.005. Epub 2013 Mar 26. J Clin Lipidol. 2013. PMID: 23642322 Free PMC article.
-
Serum lipids, plant sterols, and cholesterol kinetic responses to plant sterol supplementation in phytosterolemia heterozygotes and control individuals.Am J Clin Nutr. 2012 Apr;95(4):837-44. doi: 10.3945/ajcn.111.028985. Epub 2012 Feb 29. Am J Clin Nutr. 2012. PMID: 22378727 Free PMC article. Clinical Trial.
References
-
- Gould RG, Jones RJ, LeRoy GV, et al. Absorbability of beta-sitosterol in humans. Metabolism. 1969;18:652–662. - PubMed
-
- Salen G, Tint GS, Shefer S, et al. Increased sitosterol absorption is offset by rapid elimination to prevent accumulation in heterozygotes with sitosterolemia. Arterioscler Thromb. 1992;12:563–568. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
