

Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia
- PMID: 11179008
- PMCID: PMC1274473
- DOI: 10.1086/318810
Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia
Abstract
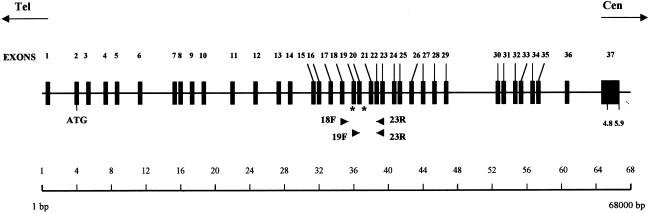
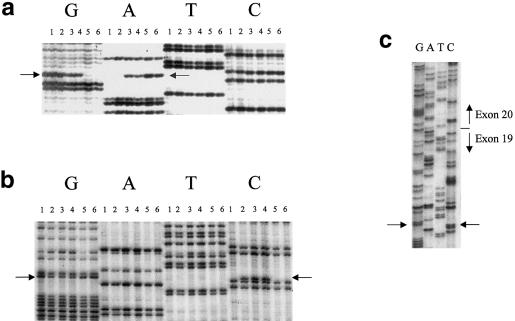
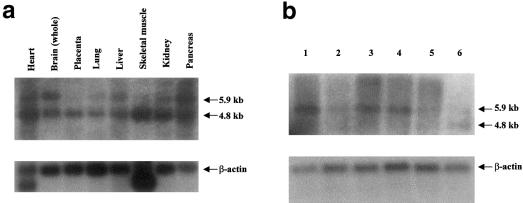
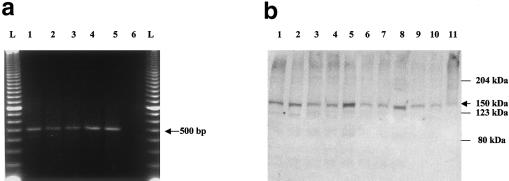
Familial dysautonomia (FD; also known as "Riley-Day syndrome"), an Ashkenazi Jewish disorder, is the best known and most frequent of a group of congenital sensory neuropathies and is characterized by widespread sensory and variable autonomic dysfunction. Previously, we had mapped the FD gene, DYS, to a 0.5-cM region on chromosome 9q31 and had shown that the ethnic bias is due to a founder effect, with >99.5% of disease alleles sharing a common ancestral haplotype. To investigate the molecular basis of FD, we sequenced the minimal candidate region and cloned and characterized its five genes. One of these, IKBKAP, harbors two mutations that can cause FD. The major haplotype mutation is located in the donor splice site of intron 20. This mutation can result in skipping of exon 20 in the mRNA of patients with FD, although they continue to express varying levels of wild-type message in a tissue-specific manner. RNA isolated from lymphoblasts of patients is primarily wild-type, whereas only the deleted message is seen in RNA isolated from brain. The mutation associated with the minor haplotype in four patients is a missense (R696P) mutation in exon 19, which is predicted to disrupt a potential phosphorylation site. Our findings indicate that almost all cases of FD are caused by an unusual splice defect that displays tissue-specific expression; and they also provide the basis for rapid carrier screening in the Ashkenazi Jewish population.
Figures
Similar articles
-
Tissue-specific reduction in splicing efficiency of IKBKAP due to the major mutation associated with familial dysautonomia.Am J Hum Genet. 2003 Mar;72(3):749-58. doi: 10.1086/368263. Epub 2003 Feb 6. Am J Hum Genet. 2003. PMID: 12577200 Free PMC article.
-
Familial dysautonomia is caused by mutations of the IKAP gene.Am J Hum Genet. 2001 Mar;68(3):753-8. doi: 10.1086/318808. Epub 2001 Jan 22. Am J Hum Genet. 2001. PMID: 11179021 Free PMC article.
-
Familial dysautonomia: detection of the IKBKAP IVS20(+6T --> C) and R696P mutations and frequencies among Ashkenazi Jews.Am J Med Genet. 2002 Jul 1;110(3):253-7. doi: 10.1002/ajmg.10450. Am J Med Genet. 2002. PMID: 12116234
-
The molecular basis of familial dysautonomia: overview, new discoveries and implications for directed therapies.Neuromolecular Med. 2008;10(3):148-56. doi: 10.1007/s12017-007-8019-5. Epub 2007 Nov 6. Neuromolecular Med. 2008. PMID: 17985250 Review.
-
Familial dysautonomia.Curr Opin Genet Dev. 2002 Jun;12(3):307-11. doi: 10.1016/s0959-437x(02)00303-9. Curr Opin Genet Dev. 2002. PMID: 12076674 Review.
Cited by
-
DERP6 (ELP5) and C3ORF75 (ELP6) regulate tumorigenicity and migration of melanoma cells as subunits of Elongator.J Biol Chem. 2012 Sep 21;287(39):32535-45. doi: 10.1074/jbc.M112.402727. Epub 2012 Aug 1. J Biol Chem. 2012. PMID: 22854966 Free PMC article.
-
Structural basis for tRNA modification by Elp3 from Dehalococcoides mccartyi.Nat Struct Mol Biol. 2016 Sep;23(9):794-802. doi: 10.1038/nsmb.3265. Epub 2016 Jul 25. Nat Struct Mol Biol. 2016. PMID: 27455459 Free PMC article.
-
Cardiac-locked bursts of muscle sympathetic nerve activity are absent in familial dysautonomia.J Physiol. 2013 Feb 1;591(3):689-700. doi: 10.1113/jphysiol.2012.246264. Epub 2012 Nov 19. J Physiol. 2013. PMID: 23165765 Free PMC article.
-
Pacemakers in patients with familial dysautonomia--a review of experience with 20 patients.Clin Auton Res. 2005 Feb;15(1):15-20. doi: 10.1007/s10286-005-0218-2. Clin Auton Res. 2005. PMID: 15768197 Review.
-
Limited complementarity between U1 snRNA and a retroviral 5' splice site permits its attenuation via RNA secondary structure.Nucleic Acids Res. 2009 Dec;37(22):7429-40. doi: 10.1093/nar/gkp694. Nucleic Acids Res. 2009. PMID: 19854941 Free PMC article.
References
Electronic-Database Information
-
- FlyBase, http://flybase.bio.indiana.edu/
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank (for published IKBKAP sequence [accession number AF044195] and 5.9-kb IKBKAP cDNA sequence [accession number AF153419])
-
- GENSCAN Web Server, http://genes.mit.edu/GENSCAN.html
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for FD (MIM 223900])
References
-
- Axelrod FB (1984) Familial dysautonomia and other congenital and sensory autonomic neuropathies. In: Blake IB (ed) Cell and molecular biology of neuronal development. Plenum Press, New York, pp 331–340
-
- ——— (1995) Familial dysautonomia. In: Robertson D, Biaggioni I (eds) Disorders of the autonomic nervous system. Harwood Academic, Luxembourg, pp 217–231
-
- ——— (1996) Familial dysautonomia. In: Robertson D, Low PA, Polinsky RJ (eds) Primer on the autonomic nervous system. Academic Press, San Diego, pp 242–249
-
- Axelrod FB, Nachtigal R, Dancis J (1974) Familial dysautonomia: diagnosis, pathogenesis and management. Adv Pediatr 21:75–96 - PubMed
-
- Axelrod FB, Pearson J (1984) Congenital sensory neuropathies. Diagnostic distinction from familial dysautonomia. Am J Dis Child 138:947–954 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
