

Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current
- PMID: 10944223
- PMCID: PMC16902
- DOI: 10.1073/pnas.97.17.9549
Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current
Abstract
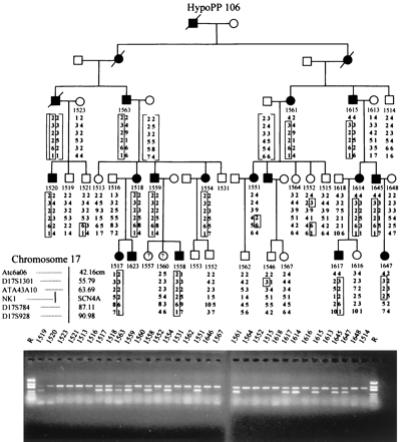
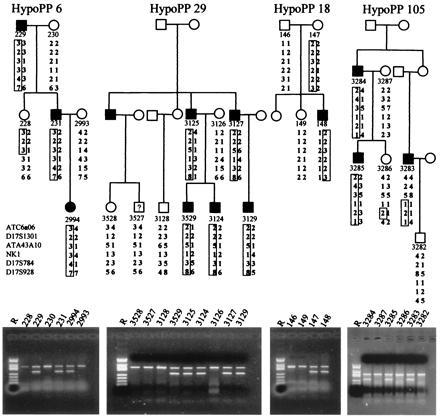
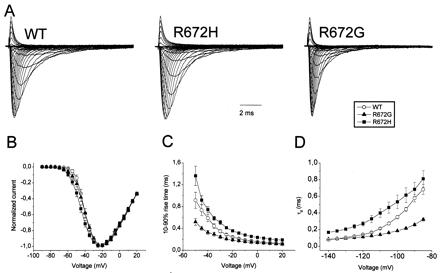
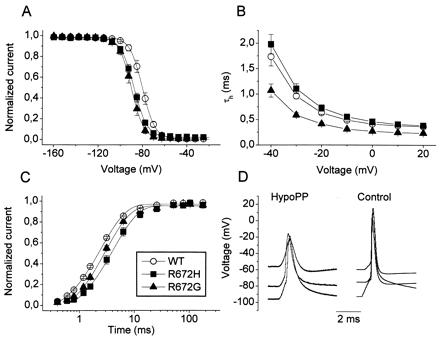
The pathomechanism of familial hypokalemic periodic paralysis (HypoPP) is a mystery, despite knowledge of the underlying dominant point mutations in the dihydropyridine receptor (DHPR) voltage sensor. In five HypoPP families without DHPR gene defects, we identified two mutations, Arg-672-->His and -->Gly, in the voltage sensor of domain 2 of a different protein: the skeletal muscle sodium channel alpha subunit, known to be responsible for hereditary muscle diseases associated with myotonia. Excised skeletal muscle fibers from a patient heterozygous for Arg-672-->Gly displayed depolarization and weakness in low-potassium extracellular solution. Slowing and smaller size of action potentials were suggestive of excitability of the wild-type channel population only. Heterologous expression of the two sodium channel mutations revealed a 10-mV left shift of the steady-state fast inactivation curve enhancing inactivation and a sodium current density that was reduced even at potentials at which inactivation was removed. Decreased current and small action potentials suggested a low channel protein density. The alterations are decisive for the pathogenesis of episodic muscle weakness by reducing the number of excitable sodium channels particularly at sustained membrane depolarization. The results prove that SCN4A, the gene encoding the sodium channel alpha subunit of skeletal muscle is responsible for HypoPP-2 which does not differ clinically from DHPR-HypoPP. HypoPP-2 represents a disease caused by enhanced channel inactivation and current reduction showing no myotonia.
Figures
Comment in
-
Skeletal muscle sodium current is reduced in hypokalemic periodic paralysis.Proc Natl Acad Sci U S A. 2000 Aug 29;97(18):9832-3. doi: 10.1073/pnas.170293197. Proc Natl Acad Sci U S A. 2000. PMID: 10954743 Free PMC article. Review. No abstract available.
Similar articles
-
The human skeletal muscle Na channel mutation R669H associated with hypokalemic periodic paralysis enhances slow inactivation.J Neurosci. 2000 Dec 1;20(23):8610-7. doi: 10.1523/JNEUROSCI.20-23-08610.2000. J Neurosci. 2000. PMID: 11102465 Free PMC article.
-
Enhanced inactivation and pH sensitivity of Na(+) channel mutations causing hypokalaemic periodic paralysis type II.Brain. 2002 Apr;125(Pt 4):835-43. doi: 10.1093/brain/awf071. Brain. 2002. PMID: 11912116
-
Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A.Brain. 2001 Jun;124(Pt 6):1091-9. doi: 10.1093/brain/124.6.1091. Brain. 2001. PMID: 11353725
-
Sodium channelopathies of skeletal muscle result from gain or loss of function.Pflugers Arch. 2010 Jul;460(2):239-48. doi: 10.1007/s00424-010-0814-4. Epub 2010 Mar 17. Pflugers Arch. 2010. PMID: 20237798 Free PMC article. Review.
-
[From gene to diseases; hypokalemic periodic paralysis].Ned Tijdschr Geneeskd. 2004 May 22;148(21):1035-8. Ned Tijdschr Geneeskd. 2004. PMID: 15185439 Review. Dutch.
Cited by
-
A calcium channel mutant mouse model of hypokalemic periodic paralysis.J Clin Invest. 2012 Dec;122(12):4580-91. doi: 10.1172/JCI66091. Epub 2012 Nov 26. J Clin Invest. 2012. PMID: 23187123 Free PMC article.
-
Multiple pore conformations driven by asynchronous movements of voltage sensors in a eukaryotic sodium channel.Nat Commun. 2013;4:1350. doi: 10.1038/ncomms2356. Nat Commun. 2013. PMID: 23322038 Free PMC article.
-
Domain IV voltage-sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels.J Gen Physiol. 2013 Aug;142(2):101-12. doi: 10.1085/jgp.201310998. Epub 2013 Jul 15. J Gen Physiol. 2013. PMID: 23858005 Free PMC article.
-
Slow inactivation does not block the aqueous accessibility to the outer pore of voltage-gated Na channels.J Gen Physiol. 2002 Oct;120(4):509-16. doi: 10.1085/jgp.20028672. J Gen Physiol. 2002. PMID: 12356853 Free PMC article.
-
Leaky sodium channels from voltage sensor mutations in periodic paralysis, but not paramyotonia.Neurology. 2011 May 10;76(19):1635-41. doi: 10.1212/WNL.0b013e318219fb57. Epub 2011 Apr 13. Neurology. 2011. PMID: 21490317 Free PMC article.
References
-
- Lehmann-Horn F, Engel A, Rüdel R, Ricker K. In: Myology. Engel A G, Franzini-Armstrong C, editors. New York: McGraw-Hill; 1994. pp. 1304–1334.
-
- Fontaine B, Khurana T S, Hoffman E P, Bruns G A, Haines J L, Trofatter J A, Hanson M P, Rich J, McFarlane H, McKenna-Yasek D, et al. Science. 1990;250:1000–1003. - PubMed
-
- Rojas C V, Wang J Z, Schwartz L S, Hoffman E P, Powell B R, Brown R H., Jr Nature (London) 1991;354:387–389. - PubMed
-
- Lehmann-Horn F, Jurkat-Rott K. Physiol Rev. 1999;79:1317–1371. - PubMed
-
- Lehmann-Horn F, Küther G, Ricker K, Grafe P, Ballanyi K, Rüdel R. Muscle Nerve. 1987;10:363–374. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
