

A double mutation in families with periodic paralysis defines new aspects of sodium channel slow inactivation
- PMID: 10930446
- PMCID: PMC314328
- DOI: 10.1172/JCI9654
A double mutation in families with periodic paralysis defines new aspects of sodium channel slow inactivation
Abstract
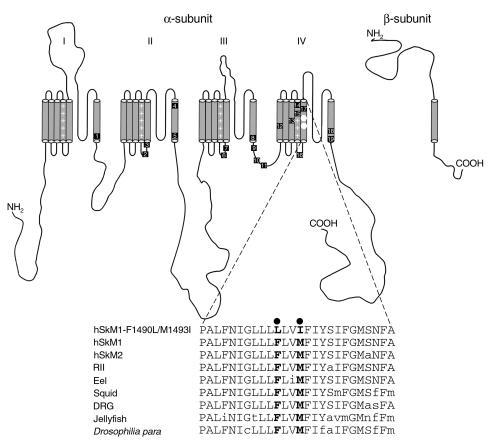
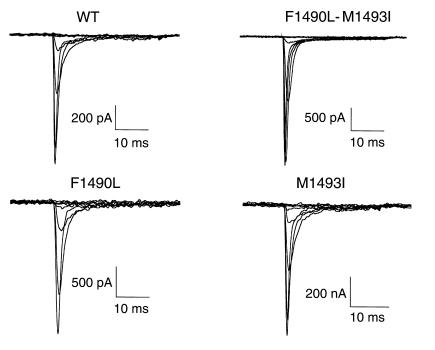
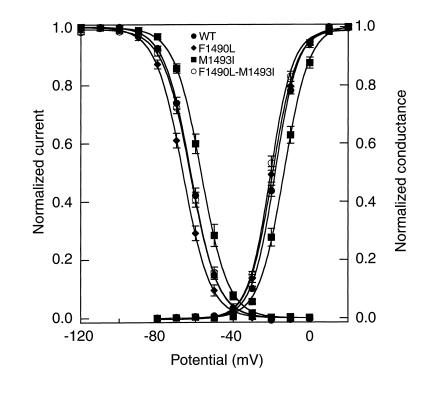
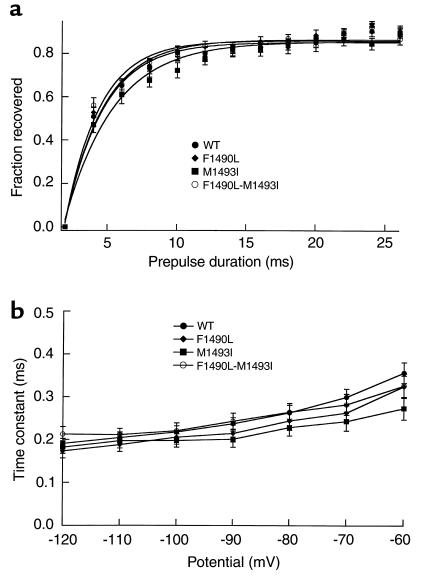
Hyperkalemic periodic paralysis (HyperKPP) is an autosomal dominant skeletal muscle disorder caused by single mutations in the SCN4A gene, encoding the human skeletal muscle voltage-gated Na(+) channel. We have now identified one allele with two novel mutations occurring simultaneously in the SCN4A gene. These mutations are found in two distinct families that had symptoms of periodic paralysis and malignant hyperthermia susceptibility. The two nucleotide transitions predict phenylalanine 1490-->leucine and methionine 1493-->isoleucine changes located in the transmembrane segment S5 in the fourth repeat of the alpha-subunit Na(+) channel. Surprisingly, this mutation did not affect fast inactivation parameters. The only defect produced by the double mutant (F1490L-M1493I, expressed in human embryonic kidney 293 cells) is an enhancement of slow inactivation, a unique behavior not seen in the 24 other disease-causing mutations. The behavior observed in these mutant channels demonstrates that manifestation of HyperKPP does not necessarily require disruption of slow inactivation. Our findings may also shed light on the molecular determinants and mechanism of Na(+) channel slow inactivation and help clarify the relationship between Na(+) channel defects and the long-term paralytic attacks experienced by patients with HyperKPP.
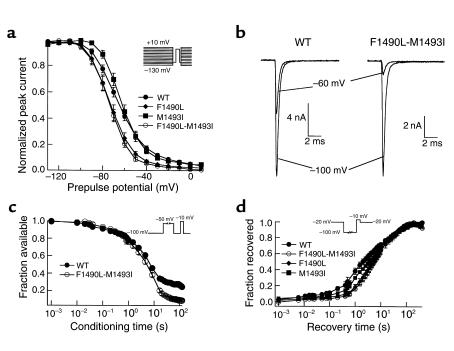
Figures
Similar articles
-
Linkage of malignant hyperthermia and hyperkalemic periodic paralysis to the adult skeletal muscle sodium channel (SCN4A) gene in a large pedigree.Am J Med Genet. 1998 Feb 26;76(1):21-7. Am J Med Genet. 1998. PMID: 9508059
-
Slow inactivation differs among mutant Na channels associated with myotonia and periodic paralysis.Biophys J. 1997 Mar;72(3):1204-19. doi: 10.1016/S0006-3495(97)78768-X. Biophys J. 1997. PMID: 9138567 Free PMC article.
-
A C-terminal skeletal muscle sodium channel mutation associated with myotonia disrupts fast inactivation.J Physiol. 2005 Jun 1;565(Pt 2):371-80. doi: 10.1113/jphysiol.2005.082909. Epub 2005 Mar 17. J Physiol. 2005. PMID: 15774523 Free PMC article.
-
Spectrum of sodium channel disturbances in the nondystrophic myotonias and periodic paralyses.Kidney Int. 2000 Mar;57(3):772-9. doi: 10.1046/j.1523-1755.2000.00914.x. Kidney Int. 2000. PMID: 10720928 Review.
-
[Familial hyperkalemic periodic paralysis: a brief review of the adult human skeletal muscle sodium channel and the application of LA-PCR to the SCN4A gene analysis].Nihon Rinsho. 1997 Dec;55(12):3253-8. Nihon Rinsho. 1997. PMID: 9436446 Review. Japanese.
Cited by
-
Inherited disorders of voltage-gated sodium channels.J Clin Invest. 2005 Aug;115(8):1990-9. doi: 10.1172/JCI25505. J Clin Invest. 2005. PMID: 16075039 Free PMC article. Review.
-
Substitutions of the S4DIV R2 residue (R1451) in NaV1.4 lead to complex forms of paramyotonia congenita and periodic paralyses.Sci Rep. 2018 Feb 1;8(1):2041. doi: 10.1038/s41598-018-20468-0. Sci Rep. 2018. PMID: 29391559 Free PMC article.
-
Novel mutations in human and mouse SCN4A implicate AMPK in myotonia and periodic paralysis.Brain. 2014 Dec;137(Pt 12):3171-85. doi: 10.1093/brain/awu292. Epub 2014 Oct 27. Brain. 2014. PMID: 25348630 Free PMC article.
-
Residue-specific effects on slow inactivation at V787 in D2-S6 of Na(v)1.4 sodium channels.Biophys J. 2001 Oct;81(4):2100-11. doi: 10.1016/S0006-3495(01)75858-4. Biophys J. 2001. PMID: 11566781 Free PMC article.
-
Skeletal muscle na channel disorders.Front Pharmacol. 2011 Oct 14;2:63. doi: 10.3389/fphar.2011.00063. eCollection 2011. Front Pharmacol. 2011. PMID: 22016737 Free PMC article.
References
-
- Ptácek, L.J., and Griggs, R.C. 1996. Familial periodic paralysis. Plenum Press. New York, New York, USA. 625–642.
-
- Bulman DE. Phenotype variation and newcomers in ion channel disorders. Hum Mol Genet. 1997;10:1679–1685. - PubMed
-
- Ptácek LJ, et al. Identification of a mutation in the gene causing hyperkalemic periodic paralysis. Cell. 1991;67:1021–1027. - PubMed
-
- Rojas CV, et al. A Met-to-Val mutation in the skeletal muscle Na+ channel α-subunit in hyperkalaemic periodic paralysis. Nature. 1991;354:387–389. - PubMed
-
- McClatchey AI, et al. Novel mutations in families with unusual and variable disorders of skeletal muscle sodium channel. Nat Genet. 1992;2:148–152. - PubMed
