

Mapping of a gene determining familial partial epilepsy with variable foci to chromosome 22q11-q12
- PMID: 10577924
- PMCID: PMC1288381
- DOI: 10.1086/302649
Mapping of a gene determining familial partial epilepsy with variable foci to chromosome 22q11-q12
Abstract
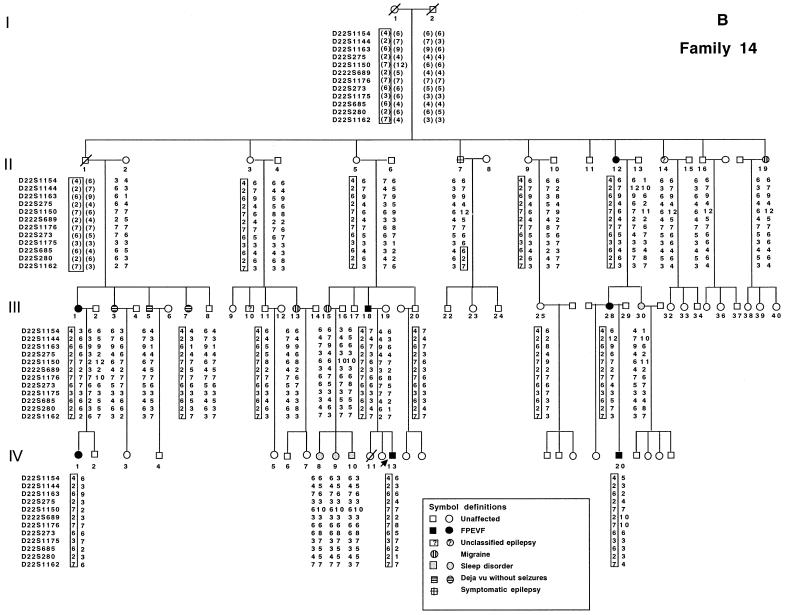
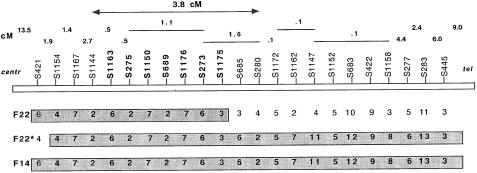
We identified two large French-Canadian families segregating a familial partial epilepsy syndrome with variable foci (FPEVF) characterized by mostly nocturnal seizures arising from frontal, temporal, and occasionally occipital epileptic foci. There is no evidence for structural brain damage or permanent neurological dysfunction. The syndrome is inherited as an autosomal dominant trait with incomplete penetrance. We mapped the disease locus to a 3. 8-cM interval on chromosome 22q11-q12, between markers D22S1144 and D22S685. Using the most conservative diagnostic scheme, the maximum cumulative LOD score was 6.53 at recombination fraction (straight theta) 0 with D22S689. The LOD score in the larger family was 5.34 at straight theta=0 with the same marker. The two families share an identical linked haplotype for >/=10 cM, including the candidate interval, indicating a recent founder effect. A severe phenotype in one of the probands may be caused by homozygosity for the causative mutation, as suggested by extensive homozygosity for the linked haplotype and a bilineal family history of epilepsy. An Australian family with a similar phenotype was not found to link to chromosome 22, indicating genetic heterogeneity of FPEVF.
Figures

Similar articles
-
Familial partial epilepsy with variable foci: clinical features and linkage to chromosome 22q12.Epilepsia. 2004 Sep;45(9):1054-60. doi: 10.1111/j.0013-9580.2004.30502.x. Epilepsia. 2004. PMID: 15329069
-
Familial partial epilepsy with variable foci in a Dutch family: clinical characteristics and confirmation of linkage to chromosome 22q.Epilepsia. 2003 Oct;44(10):1298-305. doi: 10.1046/j.1528-1157.2003.62302.x. Epilepsia. 2003. PMID: 14510823
-
Familial partial epilepsy with variable foci: a new family with suggestion of linkage to chromosome 22q12.Epilepsia. 2010 Sep;51(9):1910-4. doi: 10.1111/j.1528-1167.2010.02680.x. Epilepsia. 2010. PMID: 20659149
-
Benign adult familial myoclonic epilepsy (BAFME): evidence of an extended founder haplotype on chromosome 2p11.1-q12.2 in five Italian families.Neurogenetics. 2008 May;9(2):139-42. doi: 10.1007/s10048-008-0118-4. Epub 2008 Jan 30. Neurogenetics. 2008. PMID: 18231815
-
Genetic focal epilepsies: state of the art and paths to the future.Epilepsia. 2005;46 Suppl 10:61-7. doi: 10.1111/j.1528-1167.2005.00361.x. Epilepsia. 2005. PMID: 16359475 Review.
Cited by
-
Genetic basis in epilepsies caused by malformations of cortical development and in those with structurally normal brain.Hum Genet. 2009 Jul;126(1):173-93. doi: 10.1007/s00439-009-0702-1. Epub 2009 Jun 18. Hum Genet. 2009. PMID: 19536565 Review.
-
Identification of epilepsy genes in human and mouse.Annu Rev Genet. 2001;35:567-88. doi: 10.1146/annurev.genet.35.102401.091142. Annu Rev Genet. 2001. PMID: 11700294 Free PMC article. Review.
-
Epileptic spasms are a feature of DEPDC5 mTORopathy.Neurol Genet. 2015 Jul 23;1(2):e17. doi: 10.1212/NXG.0000000000000016. eCollection 2015 Aug. Neurol Genet. 2015. PMID: 27066554 Free PMC article.
-
Progress in the genetics of the partial epilepsies.Epilepsia. 2001;42 Suppl 5(Suppl 5):24-30. doi: 10.1046/j.1528-1157.2001.0420s5024.x. Epilepsia. 2001. PMID: 11887964 Free PMC article. Review.
-
A new locus for autosomal dominant intracranial aneurysm, ANIB4, maps to chromosome 5p15.2-14.3.J Med Genet. 2006 Jun;43(6):e31. doi: 10.1136/jmg.2005.033209. J Med Genet. 2006. PMID: 16740915 Free PMC article.
References
Electronic-Database Information
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
-
- GeneMap 99, http://www.ncbi.nlm.nih.gov/genemap99/
-
- Généthon map, ftp://ftp.genethon.fr/pub/Gmap/Nature-1995
-
- Genetic Location Database, ftp://cedar.genetics.soton.ac.uk/public_html/index.html
-
- Sanger Centre map of chromosome 22, http://www.sanger.ac.uk/HGP/Chr22/
References
-
- Andermann E, Abou-Khalil A, Berkovic SF, Javidan M, Fish D, Pandolfo M, Andermann F (1997) Deja-vu is the characteristic aura in benign familial temporal lobe epilepsy. Epilepsia 38:200 - PubMed
-
- Berkovic SF, McIntosh A, Howell RA, Mitchell A, Sheffield LJ, Hopper JL (1996) Familial temporal lobe epilepsy: a common disorder identified in twins. Ann Neurol 40:227–235 - PubMed
-
- Berkovic SF, Scheffer IE (1997a) Epilepsies with single gene inheritance. Brain Dev 19:13–18 - PubMed
-
- ——— (1997b) Genetics of human partial epilepsy. Curr Opin Neurol 10:110–114 - PubMed
-
- Cendes F, Lopes-Cendes I, Andermann E, Andermann F (1998) Familial temporal lobe epilepsy: a clinically heterogeneous syndrome. Neurology 50:554–557 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
